제목: scGPT: toward building a foundation model for single-cell multi-omics using generative AI
출판: Nature Methods(2024년 2월)
키워드: scGPT, foundation model, generative pretraining
중요도
Introduction, Abstract > Methods > Discussion > (Background) > Results
Abstract
Foundation model을 개발함에 있어서 large-scale diverse datasets + pretrained transformer의 조합이 등장하였다. 이 조합은 세포생물학에서도 사용될 수 있는데, 그 이유는 언어에서 text가 word의 집합인 것처럼, cell도 gene들의 집합으로 정의될 수 있기 때문이다. 본 연구진들은 scGPT라는 single-cell biology의 foundation model을 개발했고, 이는 3300만개의 세포 데이터를 이용해 pretrained된 transformer기반의 생성형 모델이다. 이 모델은 유전자, 세포에 대한 중요한 생물학적 인사이트를 압축해서 담고 있음을 확인하였고, 다양한 downstream tasks(cell type annotation, multi-batch integration, multil-omic integration...)에서 뛰어난 성능을 보였다.
Introduction
scRNA-seq은 다양한 세포를 정밀 분석해 질병 기전을 밝히고 맞춤 치료를 가능하게 하며, 수천만 세포를 포함한 인간 세포 지도가 구축되는 기반이 되었다. 최근 시퀀싱 기술의 발전으로 다양한 데이터 모달리티가 생성되면서, genomics를 넘어 후성유전체, 전사체, 단백질체까지 이해가 확장되었고, 이를 통한 multi-modal 분석이 가능해졌다. 이에 따라 reference mapping, perturbation prediction, multi-omic integration과 같은 새로운 연구 질문들이 생겨났고, 방법론 개발이 중요해졌다.
대표적인 방법은 foundation model들의 generative pretraining이다. 주로 self-attention transformer 아키텍쳐 기반인 것들이 많고, 예시로 Enformer가 있다. 이러한 foundation model들은 task-specific model보다 성능이 뛰어나서, single-cell omics에도 적용을 하려는 시도로 이어졌다.
머신러닝 모델들이 존재하기는 하지만, 일반적인 모델이 아니라, task-specific model이어서, 데이터셋이 스케일적으로 한계가 있는 경우에는 적용하기가 어렵다. 이러한 한계를 극복하기 위해 대규모 데이터셋에서 pretrained된 foundation model의 필요성이 대두되었다. Foundation model은 다양한 조직들에서 유전자간의 복잡한 상호작용을 이해할 수 있을 것이라고 기대된다.
single-cell sequencing data 모델링을 위해, 자연어생성 분야의 self-supervised pretraining workflow를 참고하였다.
self-attention transformer는 다음과 같은 두 가지 주요 특징이 있다.
- input token modeling
- input token의 유연성; 추가적인 feature, meta-information 포함 가능
첫번째로는, 자연어생성 분야에서 text가 word로 이루어진 것처럼, cell을 gene, protein(유전자로부터 인코딩된)으로 특징지어질 수 있다. 따라서 유전자, 세포 임베딩을 각각 학습하는 것이 아니라, 동시에 학습함으로써 세포특성을 더 잘 이해할 수 있다.
두번째 특징의 예시는 Geneformer에서 볼 수 있는데, Geneformer에서는 유전자 발현량 기준으로 유전자를 정렬함으로써 "발현량"이라는 추가적인 feature 정보를 함께 담았다.
single-cell omics data는 더 복잡하고, non-sequential 특성을 가지고 있기 때문에, 이를 반영하기 위해 pretraining workflow를 수정할 필요성이 있다.
- 자연어처럼 순서가 명확한 시퀀스 데이터는 Transformer가 잘 처리하지만, 오믹스 데이터 중에는 순서가 없거나 복잡한 관계망 구조(예: 유전자 간 상호작용, 네트워크, 조절 요소들)가 많음.
- 기존의 시퀀스 중심 사전학습 방식은 이런 비순차적, 복잡한 특성(intricacies)을 충분히 반영하지 못함.
본 연구에서는 3,300만 개 이상의 단일 세포 데이터를 이용해 사전학습한 단일세포 파운데이션 모델인 scGPT를 제시한다.
특히 non-sequential omics data를 위한 통합 generative pretraining workflow를 구축하고, 트랜스포머 구조를 세포와 유전자 표현을 동시에 학습하도록 조정하였다. 또한, 다양한 과제에 적용할 수 있도록 맞춤형 fine-tuning 파이프라인도 함께 제공한다.
scGPT 모델은 세 가지 핵심 측면에서 단일세포 파운데이션 모델의 혁신적 가능성을 보여준다.
- 대규모 생성형 파운데이션 모델로서 다양한 downstream 작업에서 뛰어난 성능을 달성하며, ‘pretraining universally, fine-tuning on demand’ 방식을 범용 솔루션으로 입증했다.
- fine-tuned된 모델과 raw 사전학습 모델 간의 유전자 임베딩과 어텐션 가중치 비교를 통해 특정 환경, 조건에서의 유전자 상호작용에 대한 중요한 생물학적 통찰을 얻었다.
- 더 큰 사전학습 데이터가 더 우수한 임베딩과 downstream 성능 향상으로 이어지는 확장 효과를 확인했으며, 이는 연구 커뮤니티의 시퀀싱 데이터 증가에 따라 파운데이션 모델이 지속 발전할 가능성을 시사한다.
Results
Single-cell transformer foundation model overview
Core model
- transformer layers with multi-head attention: generate cell & gene embeddings simultaneously
Training stage
1. Pretraining
- customized attention mask
- self-supervised 방식을 통해 세포, 유전자 표현 최적화
- learns to generate gene expression of cells
2. Fine-tuning
- 새로운 데이터셋, 특정 task에 adapt가능

Dataset
- CELLxGENE에서 얻은, healthy 33million human cells
- 51개의 장기, 조직들로부터 얻은 세포 타입을 포함한 데이터셋
- rich representation of cellular heterogeneity

pretraining 이후, scGPT cell embeddings를 10%의 인간세포(3M human cells)에서 UMAP 시각화 진행
아래 fig에서 보이는 것처럼, 명확하게 세포 타입끼리 구분되는 것을 보여주고 있다. (이 점에서 연구진들은 이 모델이 괜찮다는 확신을 얻었다고 한다.)

scGPT improves the precision of cell type annotation
neural network classifier가 scGPT에서 출력된 cell embedding을 input으로 받아서, 세포 종류에 대한 categorical predictions을 수행하여 output으로 내보낸다. reference dataset(전문가 annotation이 존재하는, 즉 supervised 형태)에서 학습을 진행한 다음, 테스트를 진행하는 방식이다.
scGPT의 cell type annotation 성능을 평가하기 위해 다양한 dataset에서 실험을 수행하였다.
human pancreas dataset
- 세포 수가 적은 희귀 세포 유형을 제외하고 대부분의 세포 유형에 대해 높은 정확도(>0.8)을 달성

- 세포 임베딩 시각화 -> 같은 세포 유형끼리는 임베딩이 가깝게 모여있음 (high intra cell type similarities)

다발성 경화증(MS, multiple sclerosis) dataset ("질병"데이터셋)
- 이번에는, 건강한 human immune cell에서 fine-tuning하고, MS condition(질병 조건)에서 예측을 평가하였다.
- 과연 건강한 데이터셋으로 fine-tuning을 해도 질병 조건하의 세포 구별을 잘 할까?!를 보고자함.
- 원래 연구에서 제공한 cell type annotation과 높은 일치율, 정확도 달성(0.85)

tumor-infiltrating myeloid dataset (면역세포 관련 데이터)
- 질병 유형간의 generalization을 위해 더 어려운 시나리오에 적용
- 6개의 암 유형에 대해 fine-tuning 후 3개의 unseen 암 유형에서 평가(fig.2d)
- immune cell subtype을 구별하는 데 높은 정확도를 보임(fig.2e,h)


위의 3가지 데이터셋에서 fine-tuning된 scGPT vs 다른 방법 비교
- 다른 최신 transformer기반 방법 TOSICA, scBERT와 비교 -> scGPT가 더 뛰어난 성능

추가로, scGPT가 cell type classification 말고도, fine-tuning없이도 unseen query cells를 reference dataset에 잘 proejct시킬 수 있는지를 확인해본 결과, 기존 방법과 비교하여 좋은 성능을 달성했음을 발견하였다.
scGPT predicts unseen genetic pertubation responses
scGPT를 활용하면, 알려진 실험에서 얻은 cellular response를 활용하여 unknown responses를 예측할 수 있다. Self-attention mechansim을 통해 pertubed genes와 다른 genes간의 복잡한 상호작용을 인코딩할 수 있기 때문이다.
Prediction of unseen gene perturbations
Perturb-seq Dataset
- Adamson dataset: 87개의 one-gene perturbations
- Replogle dataset: 1823개의 one-gene perturbations
- Norman dataset: 131개의 two-gene perturbations, 105개의 one-gene perturbations
실험 방법
- perturbations의 subset에서 fine-tuning 진행
- 대조군 cell state, 개입 유전자가 주어졌을 때 perturbed expressionon profile 예측
- unseen genes를 포함한 perturbation에 대해 test진행
Pearson delta metric
- predicted, observed post-perturbation expression changes 간의 correlation 측정
- Pearson delta on DE genes: 상위 20개의 DEGs에 대한 Pearson delta
결과
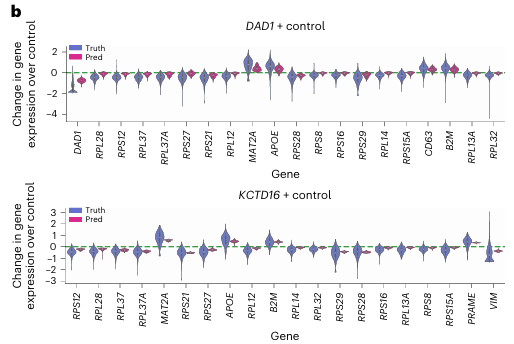
- GEARS, linear regression baseline과 비교 -> 모든 데이터셋에서 scGPT가 가장 높은 score

- Adamson dataset에서, scGPT가 두 개의 예시 perturbation에 대해, top 20 DEGs의 expression 변화 트렌드를 정확히 예측했다.
Predict unseen perturbations
- original Pertub-seq study는 236개의 perturbations targeting 105 genes를 다룸. 이들 타겟 유전자간의 조합을 모두 고려하면, 5565개의 가능한 perturbation이 있다는 소린데, 이는 곧 실제 Perturb-seq 데이터가 가능한 모든 perturbation space의 5%밖에 안된다.

- fine-tuned scGPT를 활용하여 in silico에서 각 perturbation별로 평균 response를 예측하였다. 아래는 예측한 것을 UMAP으로 시각화한 결과이다. 정답 annotation과 비교하였을 때, 같은 functional groups의 perturbation 조건이, 이웃하는 영역에 clustering됨을 발견하였다.(오른쪽 그림)


- scGPT가 예측한 발현값을 Leiden 방법을 이용해 모아보니(클러스터링) 패턴이 나타났는데, 바로 각 클러스터가 dominant gene와 강하게 연결된다는 것이다.
- dominant gene이란, 복합 교란 상황에서 발현 변화를 주도하는 주된 유전자
- 예를 들어, KLF1 유전자와 연관된 것으로 표시된 원형 클러스터는 해당 클러스터의 세포들이 KLF1과 또 다른 유전자(즉, KLF1 + X) 간의 복합 교란(perturbation)을 받은 것을 의미한다. (전부 KLF1과 다른 유전자를 함께 조작한 경우)
- KLF1과 CNN1 클러스터를 두 가지 예로 들어, 해당 클러스터에서 해당 유전자의 발현이 유독 높게 나타났음을 추가적으로 검증했다(Fig. 3e). 이는 Norman 데이터셋에서의 CRISPRa(유전자 발현을 활성화하는 CRISPR 기반 전사 활성화) Perturb-seq 실험의 예상 결과와 일치한다. → 모델이 실험 결과를 제대로 재현함
- 이러한 dominant gene 중심으로 형성된 clusters는 scGPT가 두 유전자를 동시에 조작한 경우에도 주요 유전자와 그 조합한 간의 관계, 즉 유전자 교란 조합 간의 연관성을 발견할 수 있는 능력을 보여준다.

In silico reverse perturbation prediction
scGPT는 주어진 결과 세포 상태로부터 그 상태를 유발한 perturbation의 원인을 거꾸로 예측할 수도 있다.
이런 역예측을 이상적으로 잘 수행하는 모델은 계통(lineage) 발달의 핵심 유전자를 추론하거나 치료 타겟 유전자를 발굴하는 데 유용하다.
예를 들어, 이런 기능을 활용해 질병 상태에서 회복하도록 세포를 변화시키는 CRISPR 타겟 유전자를 예측할 수 있다.
실험 설정
- 저자들은 Norman 데이터셋의 일부(20개 유전자를 포함하는 교란 데이터)에 대해 실험
- 가능한 조합은 210개 (단일 유전자 교란 + 2개 유전자 교란)
- 이 중 39개 (18%)의 알려진 교란을 학습 데이터로 사용함
- 나머지 보지 못한 세포 상태를 입력해, 그 상태를 만들 수 있는 유전자 교란 조합을 예측하게 함
결과
- scGPT는 테스트 예시 중 하나에서 CNN1 + MAPK1 교란을 1순위로 정확히 예측
- 또 다른 예시에서는 FOSB + UBASH3B 교란을 2순위로 예측
- Top-1 예측에서 평균 91.4%의 관련 교란을 찾아냄(7개 중 평균 6.4개).
- Top-8 예측에서는 평균 65.7%의 정답 교란을 찾아냄(7개 중 평균 4.6개).
- 이는 GEARS 모델이나 baseline보다 훨씬 좋은 성능
의의
- 이 방법은 교란 실험 계획에서, 목표 세포 상태를 만들 가능성이 높은 교란 조합을 우선 시도하게 해, 무작위로 시도했을 때 평균 105.5회시도가 필요한 것을 훨씬 줄일 수 있음
- 이렇게 하면 중요한 유전자 드라이버를 더 빨리 찾고, 교란 실험을 최적화할 수 있음


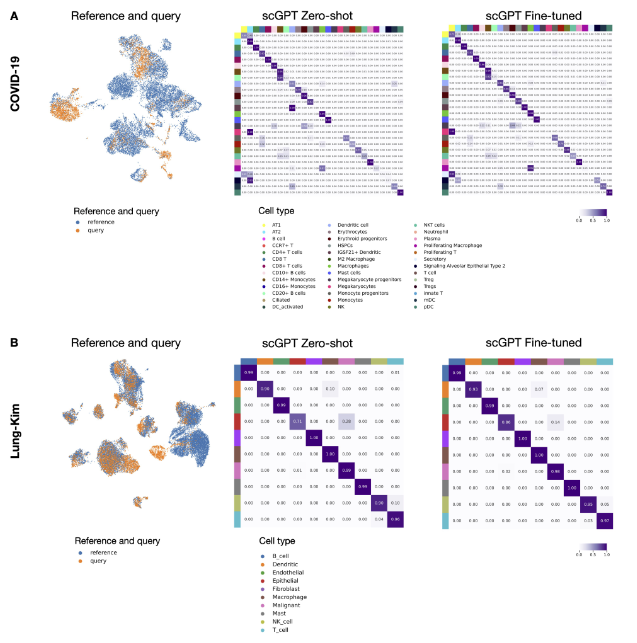
scGPT enables multi-batch and multi-omic integration
Multi-batch scRNA-seq integration
여러 배치에서 온 scRNA-seq를 통합하는 과정에서의 challenging한 점은, 생물학적인 variance를 보존함과 동시에 기술적인 배치 이펙트를 제거하는 것이다. COVID-19(18 batches) , peripheral blood mononuclear cell (PBMC) 10k (two
batches) and perirhinal cortex (two batches) datasets에서, scVI, Seurat, Harmony 방법과 비교하였다.
PBMC 10k 데이터셋에서, scGPT는 모든 세포 유형을 성공적으로 분리하였다. AvgBIO score도 0.821로, 다른 방법에 비해 높았다. scGPT는 fine-tuning 없이도 PBMC 10k datasets을 통합하는 데 상당한 성능을 보였는데, 이는 scGPT의 generalizability of pretraining을 확인할 수 있다.

한편, integration task에서 fine-tuning 작업을 가속화하기 위한 전략도 개발했는데,
(Full fine-tune (기본): 모든 파라미터 업데이트, zero-expressed gene 포함)
- Zero-expressed gene 제거: 시퀀스 길이가 40~60%로 줄어듦 → 학습 시간·GPU 사용량 약 절반 감소
- Embedding layer freeze: 임베딩 레이어 고정 → GPU 사용량 1GB 추가 절감, 학습 속도 약간 향상
cf. 임베딩 레이어를 고정하는 이유?
- 작은 downstream task에서 임베딩을 다시 학습하면 이미 학습된 의미 구조를 망칠 위험이 있음.
- 따라서 freeze하면 pre-trained knowledge를 그대로 유지할 수 있음.
그 결과,
- 속도·메모리 절감 효과 있음
- AvgBio score는 기존과 유사, 성능 손실 거의 없음
Single-cell multi-omic integration
여러 omics를 통합하는 과정에서의 challenging한 점은, biological signals를 보존하면서 cell representations를 합치는 것이다.
scGPT는 서로 다른 omics datasets으로부터 integrated cell embeddings를 효율적으로 추출함으로써 이 문제를 해결한다.
10x Multiome PBMC dataset(gene expression + chromatin accessibility)에서 scGLUE, Seurat(v.4)와 비교한 결과, scGPT가 유일하게 CD8+ naive 세포를 뚜렷하게 구분하는 cluster를 생성한 것을 확인할 수 있었다.
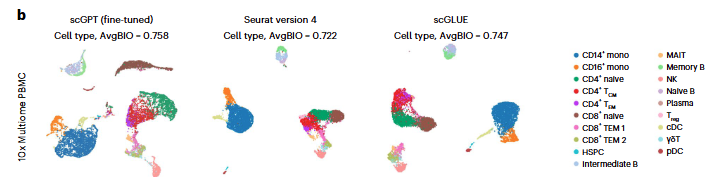
다음으로, BMMCs dataset(paired gene expression + protein abundance)에서 테스트하였는데, 이 데이터셋은 많은 양의 세포(9만개), 12 donors(12 batches), fine-grained sub-group annotations(48cell types)를 포함한다. 그 결과, scGPT가 Seruat(v.4)보다 더 뚜렷한 클러스터 구조를 보여주었다.

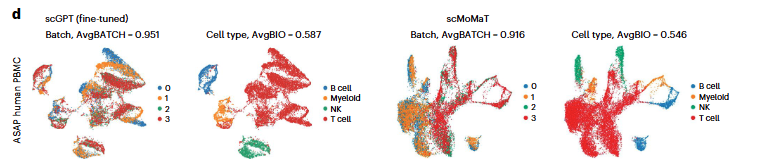
mosaic data-integration setting에서는 샘플이 모든 모달리티를 공유하지 않아 통합이 어렵다. scGPT는 ASAP PBMC 데이터셋(ATAC with select antigen profiling)(4개 배치, 3개 모달리티)에서 scMoMaT 대비 뛰어난 batch correction 성능을 보여주었으며, 특히 B세포, 골수세포, NK세포 그룹에서 효과적이었다. 전체적으로 세포 유형 클러스터링 성능이 우수하고, 다양한 생물학적 보존 지표에서도 안정적인 성능을 나타냈다.
scGPT uncovers gene networks for specific cell states
GRN에서 transcription factors, cofactors, enhancers, target genes간의 상호작용은 생물학적 프로세스를 매개하는 중요한 역할을 한다.
Existing GRN inference methods
- static gene expression에서의 상관관계에 의존
- 또는, pseudo-time(가상 시간 순서)을 인과관계를 보여주는 것처럼 간접적인 근거로 사용(인과 그래프의 추정치로 사용)
scGPT
- gene embeddings, attention maps 속에 유전자 간 관계를 암묵적으로 인코딩
- gene embeddings: gene-gene interaction을 포함하는 similarity network 구성
- attention maps: 다양한 세포 상태에서의 고유한 gene network activation patterns 포착
본 연구에서는 scGPT로부터 추출한 gene network를 기존에 알려진 생물학적 지식과 비교 검증하고, 이를 gene program 발견에 적용 가능성을 탐구한다.
사전학습된 scGPT모델에서 HLA proteins의 similarity network를 시각화한 결과(fig.5a), zero-shot setting에서, scGPT가 성공적으로 2개의 클러스터(HLA class I, class II genes)를 강조하였다. 각 클래스는 서로 다른 면역 반응 context에서 기능하는 항원제시 단백질(antigen-presenting proteins)를 인코딩한다.
추가적으로, scGPT를 immune human dataset에서 fine-tuned하고 이 데이터셋에 존재하는 면역세포 유형에 특이적인 CD 유전자 네트워크를 탐구하였다.(fig.5b)
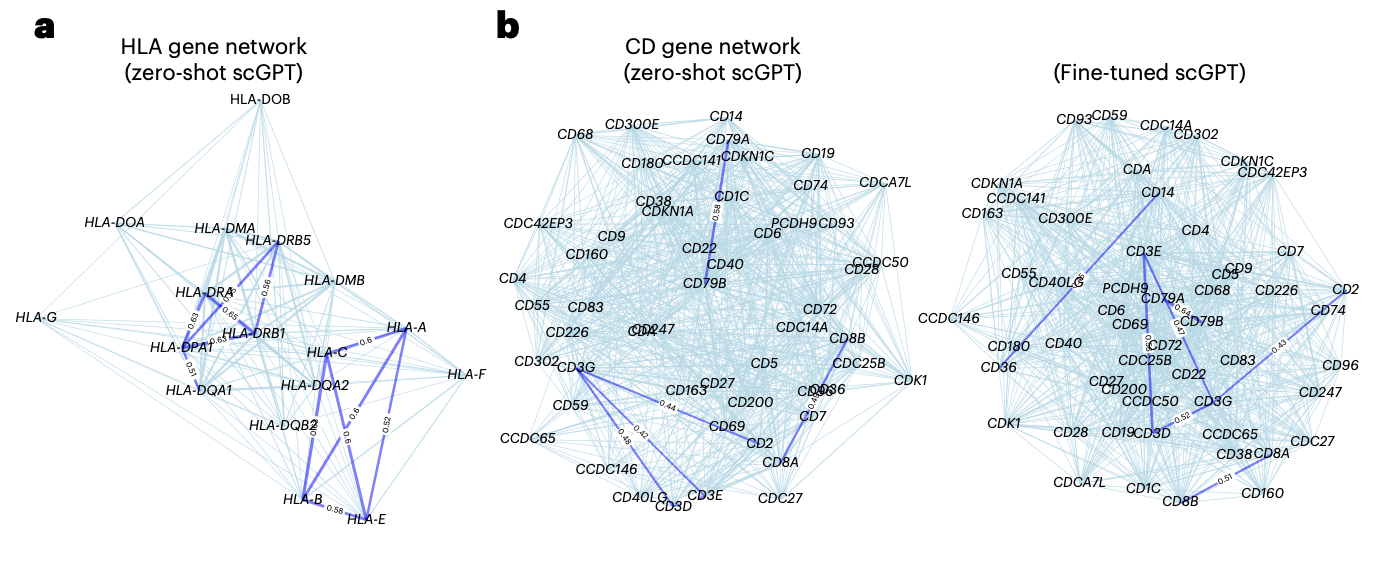
→ scGPT demonstrates its ability to group functionally related genes & differentiate functionally distinct genes
scGPT는 세포 유형에 특이적인 활성을 보이는 의미 있는 gene programs을 찾아낼 수 있다. Gene programs는 gene embeddings를 기반으로 선택되고 클러스터링되는데, fig.5c에서는 ‘immune human’ 데이터셋에서 변이가 큰 유전자들(HVGs)에 대해 fine-tuned된 scGPT가 추출한 유전자 프로그램과, 각 프로그램이 서로 다른 세포 유형에서 어떻게 발현되는지를 시각화하였다.
분석 결과, HLA class II 유전자 집합이 group 2로, T3 complex에 관여하는 CD3 유전자들이 group 3으로 확인되었으며, CD3 유전자는 T 세포에서 가장 높은 발현을 보였다.

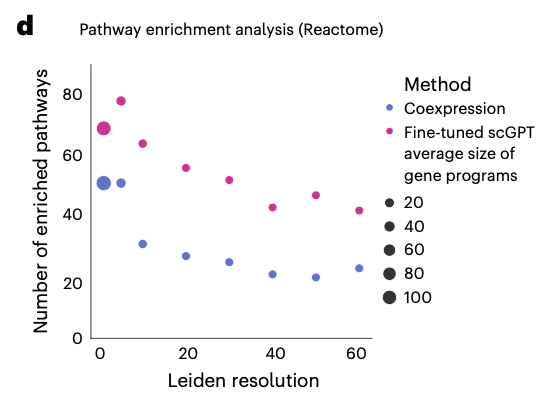
추출된 gene programs를 검증하기 위하여 pathway enrichment analysis를 진행하였다.
그 결과, scGPT가 모든 클러스터링 해상도(세밀한 정도?)에서 enriched pathways의 수가 coexpression network에 비해 상당히 더 많음이 관찰되었다. 즉, scGPT가 다양한 조건에서 더 많은 '의미있는 생물학적 경로'를 잡아낸다는 것이다. (fig. 5d)
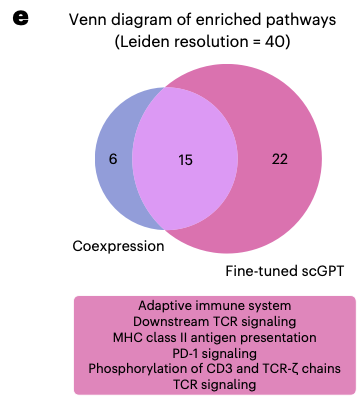
scGPT와 coexpression network간 잡아낸 pathways의 유사점, 차이점도 분석해본 결과(fig.5e), 둘다 15개의 공통된 pathway를 찾아냈고, 이는 cell cycle, immune system과 관련된 pathway이다. scGPT가 coexpression network는 못찾아낸 22개의 pathways를 더 찾아냈는데, 이 중 14개는 면역관련이었다. scGPT는 특히 adaptive immune system, T cell receptor signaling, PD-1 signaling, MHC class II presentation과 같은 pathway를 강조하였는데, 이는 fine-tuning 데이터셋에서 adaptive immune populations가 존재한 것과 일치한다.
→ scGPT는 복잡한 gene-gene connections을 잘 잡아내고, specific mechanism을 밝히는 데 탁월하다.
⁉️ 당연히 데이터셋에 adaptive immune population가 존재했기 때문에 해당 pathway를 강조할 수 밖에 없는게 아닌가..? 이게 왜 장점처럼 작성된거지
- 데이터셋에 adaptive immune population이 있다 → 모델이 adaptive immune 관련 pathway를 찾아야 한다"는 건 필수 조건이지만, 만약 모델이 이를 못 잡아냈다면, 모델의 생물학적 타당성은 의심받게 됨.
- 반대로 잘 잡아냈다는 건 모델이 실제 생물학적 맥락을 반영하고 있다는 신뢰 근거가 됨.
위에서 dataset-level에서의 gene network inference를 살펴보았는데, single-cell level에서도 어텐션 메커니즘이 gene-gene interactions을 잘 포착한다.
perturbation 실험에서, scGPT는 perturbation 이전, 이후에 각 perturbed 유전자에 의해 어떤 유전자들이 가장 많이 영향을 받았는지 gene network activation 변화를 조사하였다.

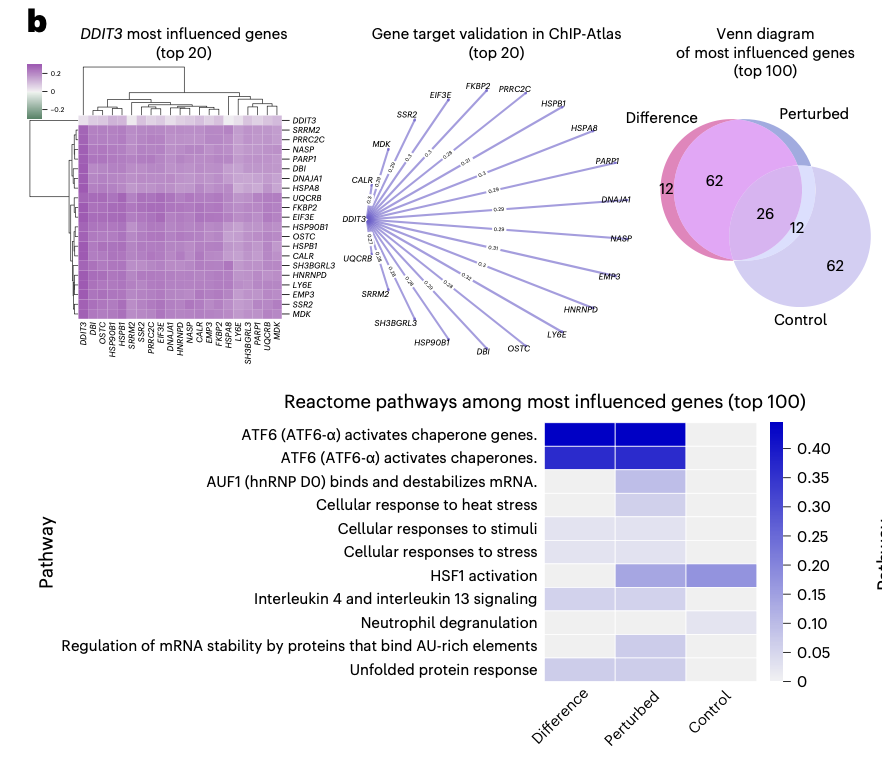
Adamson CRISPR interference dataset에서, scGPT는 DDIT3의 억제(knockout)에 의해 가장 많이 영향을 받은 top 20 유전자들을 찾아냈다. 이 유전자들은 ChIP-Atlas database에서 DDIT3의 signaling targets이다.
scGPT는 top 100 유전자들에 대해서도 pathway-activation 패턴을 찾아냈는데, 여기서 찾아진 ATF6 전사인자는 unfolded protein response를 매개하고 cell apoptosis를 조절하는 것으로 알려져있다.
cf. Difference: scGPT가 perturbed vs control 비교해서 실험적으로 가장 차이가 크다고 예측한 유전자 집합
Perturbed: scGPT가 perturbed상태에서 특히 많이 영향을 받았다고 예측한 집합
Control: scGPT가 대조군 상태에서 중요한 유전자라고 예측한 집합
BHLHE40 억제에서도 top20 영향받은 유전자들 중 19개가 BHLHE40 전사인자의 타겟으로 이미 알려진 유전자들이었다. pathway-activation profile 결과도 이 전사인자의 세포 주기 조절에서의 역할을 반영한다.
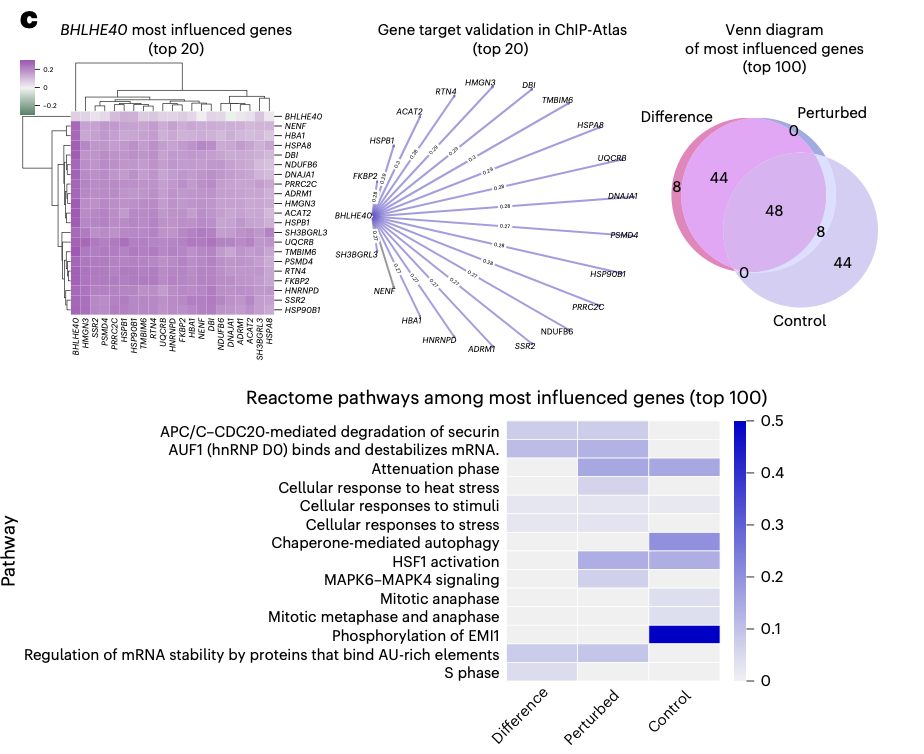
⇒ 위와 같은 attention에 기반한 발견은, scGPT가 세포 상태 수준에서 gene network를 학습했다는 것을 검증한다.
Scaling and in-context effects in transfer learning
Transfer learning process에 영향을 주는 factors를 explore해보았다.
첫번째로, pretraining data size와 fine-tuned model의 성능 간의 관계. 3M -> 33M normal human cells로 실험해본 결과, pretraining data size가 커질수록 fine-tuned model의 성능도 좋아진 것을 관찰할 수 있었다.
⇒ scaling effect
앞으로 더 크고 다양한 데이터셋이 생긴다면, 모델 성능의 향상을 기대해볼 수 있을 것이고, cellular process에 대한 이해도 향상될 수 있을 것이다.

두번째로, context-specific pretraining의 영향. 이 영향을 살펴보기 위해, 특정 세포 유형에서 사전학습을 진행하고, 비슷한 세포 유형에서 downstream task에 대해 fine-tuning을 진행하였다. 정상 인간세포를 사용하여 주요 장기별로 각각 사전학습한 7개의 organ-specific models과, 암 전반에 대한 pan-cancer 모델을 구축하였다.
cell embedding을 시각화한 결과, 아래와 같이 pan-cancer 모델의 cell embedding은 서로 다른 cancer type을 정확히 구분하는 것을 확인하였다.
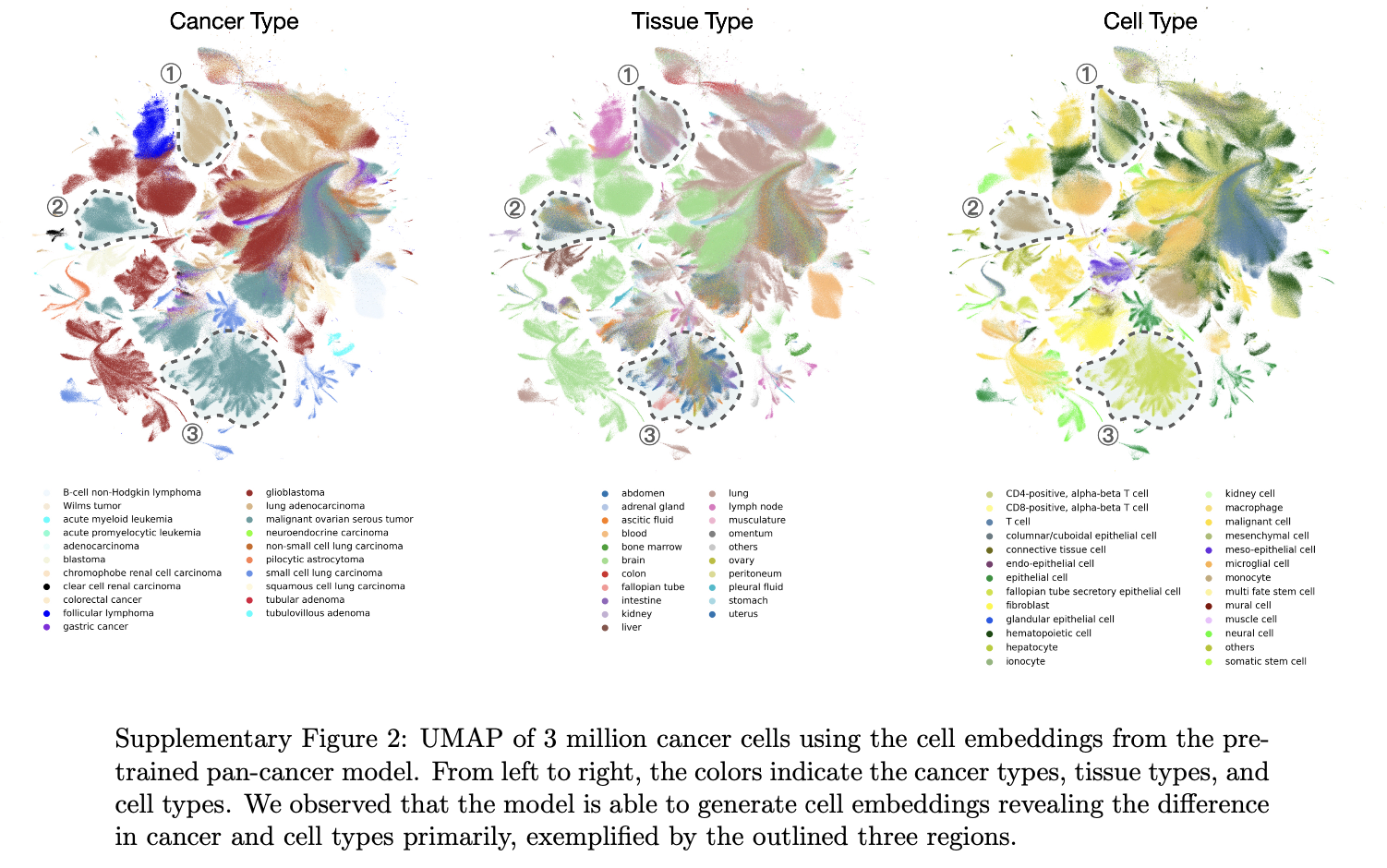
organ-specific models는 각 organs에 대해 cell heterogeneity를 잘 드러냈다. 즉, 해당 장기에 존재하는 다양한 세포 타입들을 정확하게 구분하고 각 세포의 고유 특성을 잘 반영했다.

다음으로, COVID-19 dataset에서 개별 모델을 fine-tuning하여 pretraining context의 영향을 보고자 하였는데, 그 결과, 모델의 pretraining context와 후속 데이터를 통합할 때의 성능 사이의 관련성을 확인할 수 있었다. Whole body, blood, lung dataset에서 사전학습된 모델들의 data-integration task성능이 좋았는데, 이는 COVID-19 데이터셋에 존재하는 세포 유형과 일치한다.
⇒ importance of aligning the cellular context in pretraining with the target dataset for superior results in downstream tasks 강조

Discussion
<scGPT 특징>
- foundation model; 대규모 단일세포 데이터에서 사전학습된 transformer 기반
- gene, cell embedding을 동시에 학습함 -> 다양한 세포 프로세스를 모델링 가능
- attention 메커니즘 -> 단일 세포 수준에서 유전자 간의 상호작용 포착 가능
⁉️동시에 학습한다는 의미...?? 유전자 임베딩은 word 처럼 학습하고, 유전자 임베딩을 합쳐서 세포 임베딩도 <cls>에다가 저장하기 때문에 둘다 학습한다는 뜻인듯?
<experiment>
- zero-shot experiments
- unseen datasets에 대해 추론 능력을 보임-cell type에 따른 meaningful clustering patterns 잡아냄
- fine-tuning settings
- 다양한 downstream tasks로 지식 transfer 가능 -> fine-tuned scGPT가 스크래치부터 학습된 모델들을 능가함
- 한계
- 현재의 pretraining 방식은 배치 효과(batch effect)를 자연스럽게 제거하지 못함 -> 기술적 변동이 큰 데이터셋에서는 zero-shot performance가 제한될 수 있음
- 생물학적 정답(ground truth)이 자주 없고 데이터 품질이 다양 -> 모델 평가가 복잡함
<future directions>
- 더 큰 스케일, 더 다양한 데이터셋에 pretrain을 진행할 예정
- multi-omic data, spatial omics....
- perturbation, temporal data같은 것을 pretraining에 포함
- in-context instruction learning for single-cell data 도 진행할 예정
- fine-tuning 없이, zero-shot setting에서도 다양한 테스크에 pretrained model을 바로 적용할 수 있게 해주는..
- in-context learning: 원래는 LLM 분야에서 나온 개념으로, 모델이 추가적인 재학습(fine-tuning) 없이 프롬프트 안에 주어진 “예시나 지시문(instruction)”을 참고해 새로운 문제를 푸는 학습 방식.
- 즉, 모델이 “컨텍스트(문맥)” 속에서 주어진 지시문을 이해하고, 그에 맞춰 답변/추론을 수행하는 것.
- scGPT가 다양한 분석에서 뉘앙스와 구체적인 요구조건들을 잘 캐치할 수 있게 되면, 다양한 시나리오에서의 활용성이 높아질 것.
Methods
Input embeddings
single-cell sequencing 데이터는 cell-by-gene matrix로 프로세싱 (NxG, N: # of cells, G: # of genes)
matrix에서 각 element는 특정 세포, 유전자의 RNA abundance에 해당.
Input embeddings는 아래와 같은 3개의 주요 요소로 이루어져 있다.
Gene tokens
각 유전자 g1, g2, ...에 정수 identifier id()를 assign
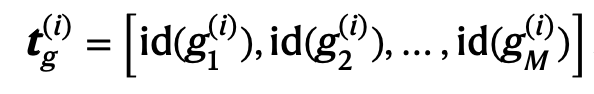
만약 서로 다른 연구(시퀀싱 기술, 전처리 파이프라인 등)에서 온 gene set이라면, 그들의 합집합을 구하는 방식으로 서로 다른 유전자 토큰 집합을 통합할 수 있다.
그리고 최종 생성된 vocabulary에 특수 토큰을 추가하는데, <cls>는 모든 유전자들을 세포 표현으로 합칠 때 사용되고, <pad>는 고정된 길이로 input을 padding할 때 사용된다.
Expression values
배치, 시퀀싱 샘플마다 data scale의 차이가 생길 수 있고, 이에 따라 같은 absolute value인데도 다른 semantic meaning을 가지고 있을수도 있다. 따라서, 이러한 차이를 극복하기 위해, value binning technique을 적용하여 expression counts를 상대적인 값으로 변환하였다. 총 B개의 intervals들이 존재하며, 모든 expression values는 특정 구간에 속하게 된다. 세포마다 bin edges를 계산했기 때문에, interval edges bk는 세포마다 다를 수 있다. 하지만, 구간 개수는 동일하다.

예를 들어, 제일 마지막 bin인 B에 속한 값들은 가장 높은 발현량을 가진 것으로 상대적으로 이해될 수 있다.
fine-tuning tasks에서는 log1p transformation, HVG selection을 먼저 진행하고 나서 value binning step을 진행하였다. 최종 binned expression value의 input vectors는 다음과 같다.

Condition tokens
condition tokens는 여러 meta 정보를 포함하고 있는데, 예를 들어 perturbation token이 있다. 각 유전자마다 condition token 값이 달라지므로, position-wise하게 condition token을 구성해야한다. 따라서, input genes와 동일한 dimension으로 input vector를 만들고, 각 integer index를 같이 표시해준다.

Embedding layers
gene tokens, condition tokens -> pytorch embedding layer (to map each token to a fixed-length embedding vecotr of dimension D)
binned expression values -> fully connected layer (to enhance expressivity)
최종 임베딩 벡터는 아래와 같다. 차원은 MxD.

Cell and gene expression modeling by transformers
scGPT transformer
self-attention transformer: encode the input embedding
input embedding: MxD에서, M개의 embedding 벡터 시퀀스에서 self-attention mechanism 진행
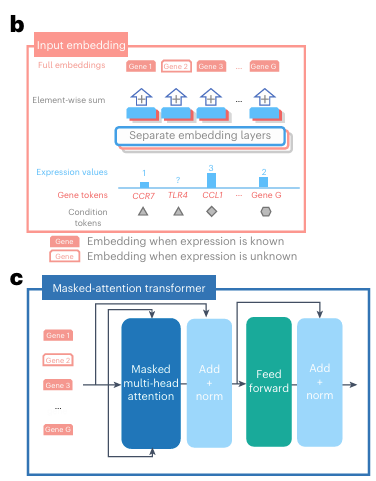
Generative pretraining
Fine-tuning objectives
Fine-tuning on downstream tasks
