제목: Transfer learning enables predictions in network biology
출판: Nature Article (2023년 5월)
요약: Geneformer
Abstract
최근, transfer learning이 대두되고 있는데, 이는 large-scale general datasets에서 pretrained된 딥러닝 모델을 downstream tasks에서 fine-tuning후 사용하는 것을 의미한다. 본 논문에서는 context-aware, attention 기반 딥러닝 모델인 Geneformer를 개발하였다. 이 모델은 3천만 single-cell transcriptomes 대규모 corpus를 기반으로 pretrained되어 데이터가 제한된 네트워크 생물학 환경에서도 context-specific predictions를 가능하게 한다. Pretraining 단계에서 Geneformer는 network dynamics에 대한 이해를 얻었고, self-supervised 방식으로 network hierarchy를 모델의 attention weights에 인코딩하였다. 여러 downstream tasks에 적용한 결과, fine-tuning을 통해 key network regulators, candidate therapeutic targets를 발견하는 과정을 가속화할 수 있을 것으로 보인다.
Introduction
질병을 타겟팅할 때, 겉으로 드러나는 결과를 타겟팅하는게 아니라, 근본적인 유전자 조절 시스템 자체를 정상화시키는 것이 더 효과적이다. 질병이 진행되는 데 핵심적인 역할을 하는 유전자 조절 네트워크(gene regulatory networks, GRN)를 파악하면, 주변부의 결과적인 요소에 집중하는 것이 아니라 핵심이 되는 조절 요소를 정상화시킬 수 있다. 즉, GRN을 파악하는 것이 중요한데, 이를 알아내기 위해서는 아주 많은 양의 transcriptomic data가 필요하다. 따라서 이러한 데이터가 부족한 희귀 질병에서는 근본적인 요소를 정상화하는 약 개발이 힘들다. 하지만, 시퀀싱 기술의 발전과, single-cell 기술의 발전으로 인해 더 많은 조직에서 RNA데이터를 수집할 수 있게 되었고, 세포 총합이 아니라 세포별 유전자 발현을 정밀하게 측정할 수 있게 되어서 가능성이 열리고 있다.
최근, transfer learning, 즉 대규모 일반 데이터셋에서 사전학습된 딥러닝 모델을, 다양한 downstream tasks에서 fine-tuning을 진행하여 사용하는 개념이 대두되었다. 이것의 장점은, 제한된 task-specific data, 즉 단독으로 사용하기에는 양이 적은 데이터도 transfer learning을 통해 유의미한 예측 결과를 얻을 수 있다는 것인데, 왜냐하면 이는 대규모의 사전학습된 근본적인 지식을 알고 있을 것이라 예상되는 딥러닝 모델을 새로운 downstream task를 해결하도록, 지식을 전달하는 개념이기 때문이다.

self-attention mechanism을 활용하면, context-aware models를 통해 각 문맥 상에서 어떤 요소에 집중해야하는지를 알 수 있게 해주고, 이는 다양한 applications에 적용가능하게 해준다. GRN 구조 자체가 굉장히 context-dependent하기 때문에(유전자간의 관계를 설명해야하므로), transformer와 같은 attention 기반 모델이 적합할 것이다.
따라서, 본 논문에서는 context-aware, attention기반의 딥러닝 모델인 Geneformer를 개발하였다. Genecorpus-30M라는 29.9million(약 3천만개) human single-cell transcriptomes포함한 데이터셋에서 pretraining을 진행하였는데, 이때 self-supervised masked learning을 통해 근본적인 네트워크에 대한 지식을 얻을 수 있게 하였다.
⁉️어떻게 self-supervised masked learning을 통해 근본적인 네트워크에 대한 지식을 얻을 수 있게 되었는가?
SSML은 특정 토큰들을 mask처리하고, 모델이 해당 부분을 예측하게 만드는 학습 방식. 아마 전사체 데이터에서는, 특정 유전자 발현값을 가리고(masking), 나머지 유전자 발현값을 통해 가려진 부분을 예측하는 방식으로 학습해서, 유전자 사이의 관계, 발현값 사이의 관계를 알아내도록 하여 관계성 패턴을 학습시키는 게 아닐까?
→ 진짜 이렇게 학습시켰는지(발현량 가리고) 코드에서 확인해보기
실제로 Geneformer를 여러 downstream tasks에 적용한 결과, predictive accuracy가 증가한 것을 관찰할 수 있었다.
이 모델은 유전자 네트워크의 핵심 조절자(예: 전사 인자, 핵심 유전자)나 약물 타겟 후보를 더 빨리 찾아내는 데 사용될 수 있다.
Geneformer architecture and pretraining
Geneformer의 핵심 특징 정리
- context-aware, self-attention
- large-scale transcriptomic data에서 사전학습됨
- network dynamics는 세포종류, 질병 상태 등에 따라 달라질 수 있는데, Geneformer의 context awareness 능력은 각 세포 context에 맞는 prediction을 가능하게 해준다.
어떻게 pretraining을 진행했는가?
<데이터 전처리>
- 대규모 사전학습 corpus인 Genecorpus-30M을 합쳤다.
- 돌연변이가 진행된 세포는 제외(malignant cells, immortalized cell lines)
- 이중 세포(doublets) 또는 손상된 세포(damaged cells)를 제외하기 위해 확장 가능한 필터링 기준(metrics) 구축
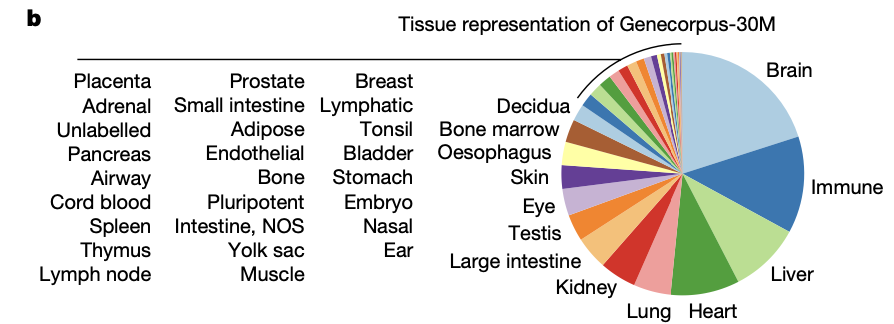
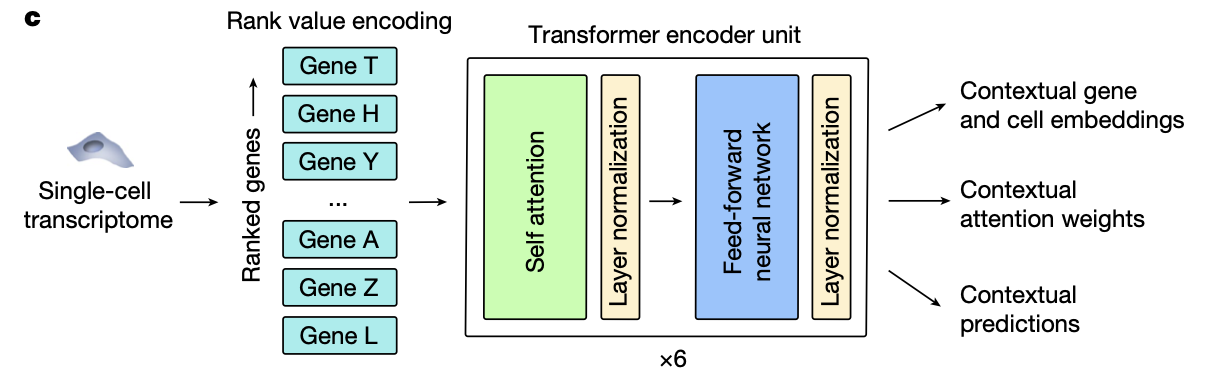
<이후의 흐름: 인코딩, transformer>
- rank value encoding
- 유전자 발현량순대로 ranked
- 절대적인 유전자 발현량 순대로 ranking하는 것이 아니라, 그 유전자의 데이터셋 전체에서의 발현량으로 정규화(비교)하여 ranking을 진행
- 따라서, 만약 특정 세포에서의 유전자 A가 전체 데이터셋에서의 유전자 A 평균 발현량보다 낮다면, 특정 세포에서 발현량이 낮다고 판단할 수 있음
- housekeeping genes(모든 세포에서 높게 발현되는 유전자)는 lower rank, transcription factors와 같이 일반적으로 낮게 발현되지만, 특정 세포에서만 높게 발현되어 세포 상태를 구별하는데 중요한 역할을 하는 유전자들은 higher rank에 매겨지게 됨.
- ⇒ 정확한 유전자 발현량을 표현할 수 없다는 단점이 있지만, 기술적 노이즈에 대해 robust하기 때문에 사용함.
- transformer
- 6개의 transformer encoder units
- 각각의 unit은 self-attention layer + feed forward neural network layer
- masked learning을 통해 사전학습 진행 (foundational knowledge에 대한 일반화 능력을 향상시키기 위해)
- 각 transcriptome마다 15%의 유전자가 masking됨, 모델은 unmasked genes의 context를 활용하여 각 masked 위치에 어떤 유전자가 있어야하는지를 예측 : self-supervised(라벨링된 데이터 필요 X)
- GPU 사용
Context awareness and batch integration
모델은 각 유전자를 256차원으로 임베딩을 진행한다. 이는 세포 맥락에 따른 유전자의 특징을 인코딩하는 것이다.
먼저, 모델이 기술적 오류(batch effects)에 영향을 받는지를 테스트하였다. 그 결과, 시퀀싱 플랫폼, 방식 등에 robust한 것을 확인할 수 있었다. 반면, 유전자 임베딩은 세포내 발현된 다른 유전자의 context에 의존적이었는데, 이는 결국 Geneformer의 context awareness를 강조한다.
예시로, 섬유아세포에 OCT4, SOX2, KLF4, MYC라는 iPSC 유도 인자를 추가했을 때, 나머지 유전자들까지 iPSC 상태로 임베딩이 이동한 것을 관찰할 수 있었다.
또한, 유전자가 매우 context-dependent한 것을 알 수 있는 예시로, GAPDH와 같은 housekeeping gene에 비해 NOTCH receptor는 임베딩이 세포마다 일정하지 않고 많이 변화하는 것을 관찰할 수 있었다.
다음으로, 유전자 임베딩을 합쳐서 세포 수준 임베딩(cell-level embeddings)을 생성하였는데, 이는 각 세포의 상태의 특징을 인코딩한 것이다. 먼저 aortic aneurysm(동맥류) 데이터로 테스트하였는데, 원래는 환자마다 발현값 차이가 커서 clustering이 엉켰는데, Geneformer의 embedding은 환자가 아니라 세포 종류/상태 중심으로 군집화되었다. 즉, 사전학습된 Geneformer가 batch effect를 극복하였음을 의미한다. 다음으로 fine-tuning하면 generalizability가 떨어지는 지 체크하였는데, Drop-seq vs DroNc-seq 플랫폼에서 각각 학습·테스트 해보니, fine-tuning을 해도 플랫폼 간 generalization 유지되고, 기존 방법(ComBat, Harmony)보다 플랫폼 간 통합이 더 잘 되는 것을 확인하였다.

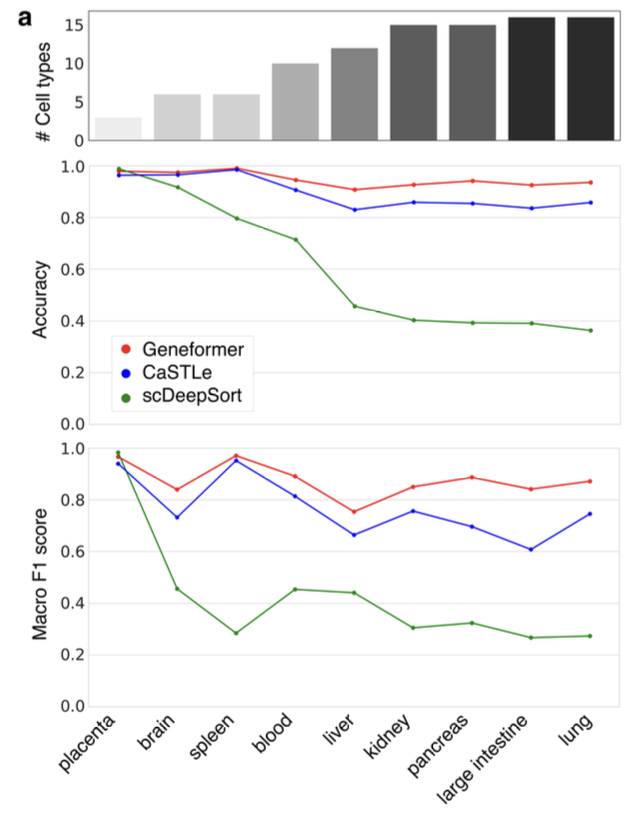
Geneformer를 cell-type annotation에서도 실험한 결과를 XGBoost와 딥러닝 기반 모델과 비교하였다. 후자는 매번 특정 조직용으로 별도로 모델을 일일이 학습해야하고(train a new model from scratch), 특정 "조직"에서의 세포 종류를 예측한다는 점에서 같은 objective을 공유하는데, Geneformer는 대규모의 라벨링되지 않은 데이터셋에서 self-supervised pretraining이 가능하기 때문에 generalizable learning objective을 통해 fundamental knowledge를 얻을 수 있다. 다른 방법과 비교하였을 때 Geneformer는 세포 종류(클래스 수)가 다양해질수록 정확도, F1 점수 모두에서 성능 우위를 보였다.
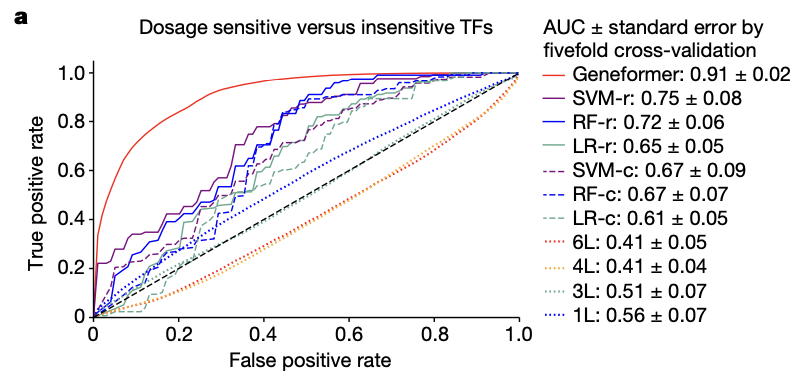
Gene dosage sensitivity predictions
복제 수 변이(CNVs)를 해석할 때의 주요 과제는, dosage-sensitive genes(유전자 복제 수의 변화에 민감한 유전자)가 어떤 것인지 파악하는 것이다. Conversation, allele frequency가 dosage-sensitivity를 예측하는데 주로 사용되긴 하지만, 이러한 특징들은 세포 상태에 따라 변화하지 않고, transcriptional dynamics를 포착할 수 없다는 한계가 있다. Dosage-sensitive VS dosage-insensitive 유전자 세트를 이용하여 Geneformer를 fine-tuning한 결과(only 10,000 random single-cell transcriptomes), 다른 방법에 비해 예측 성능이 뛰어난 것을 확인하였다. AUC=0.91
더 크고 다양한 데이터셋에서 사전학습시킨 것이 downstream task의 AUC를 더 높였다.
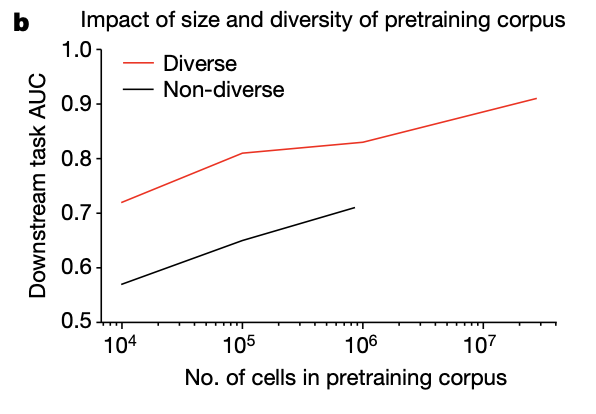
그렇다면 더이상의 학습 없이, fine-tuned된 모델이 최근에 새로 보고된 유전자들의 dosage sensitivity도 잘 예측할 수 있을까?
연구팀에서는 특정 유전자가 deletion되었을 때 신경발달질환과 관련이 있는지를 평가하고자 하였고, 그 결과를 high 또는 moderate 신뢰도로 나누어 유전자를 분류하였다. Fine-tuned된 Geneformer를 적용했을 때, fetal cerebral cells라는 context에서 high-confidence 유전자들이 dosage sensitive하다는 것을 원래 연구 결과와 96% 일치하는 수준으로 정확하게 예측했다. Moderate-confidence 유전자들에 대해서도 84%의 일치율로 해당 유전자들이 fetal cerebrum에서 dosage sensitive하다고 예측했다.
High-confidence 유전자들의 dosage-sensitivity를 다른 세포들에서도 비슷한 수준으로 예측했는데, 이는 이 유전자들이 어느 조직에서나 sensitive하다는 것을 의미한다. 반면, moderate-confidence 유전자들은 세포 타입에 따라 예측결과가 달라졌는데, 이는 context specific dosage sensitivity를 보였다는 것을 의미한다. 특히 어른의 뉴런보다 태아 뇌세포(fetal cerebrum)에서 더 높은 민감성을 보였다는 점에서, 해당 유전자들이 신경발달 질환과 관련된다는 점과 잘 들어맞는다.
⇒ Geneformer는 문맥을 인식할 수 있는 context-aware 모델!

다음으로, 어떤 유전자의 deletion이 특정 세포 문맥에서 해로운 영향을 미칠지를 예측하였다. Geneformer 모델에 입력된 유전자의 ranking 정보를 제거하여 나머지 유전자들의 embedding이 얼마나 변하는지를 측정하였다.
만약 유전자 하나를 제거했는데 나머지의 임베딩이 많이 바뀌면 이 유전자가 중요한 역할을 하고 있었다는 뜻
태아 심장 근육세포에 대해, 이미 심장질환과 관련있다고 알려진 유전자를 제거했더니, 나머지 유전자 임베딩이 흔들렸다. 즉, 제거한 유전자들이 정말 중요한 유전자라는 것이 모델로도 확인되었다. 대조군은 고지혈증 관련 유전자들인데, 이들은 심장세포에서 발현되지만 실제로 심장세포에는 영향을 주지 않는 유전자들이다. 실제로도, 이 유전자들을 제거했을 때 나머지 임베딩의 변화는 작았다.
즉, 모델의 예측이 실제 질병 연관성과 잘 맞는다는 것을 확인하였다. 그리고 새롭게 도출된 후보 유전자도 있었는데,
그 중 하나가 TEAD4라는 전사인자이다. 이전엔 dosage-sensitive 유전자로 잘 알려지지 않았지만, Geneformer는 그 중요성을 포착하였고, 이후 실험에서 해당 전사인자가 중요함을 재차 확인하였다.
Chromatin dynamics predictions
Geneformer는 단순히 유전자 발현 수준을 넘어서, 크로마틴 상태나 전사인자의 작용 거리 같은 고차원적인 유전자 조절 정보도 예측할 수 있다. 먼저, bivalent chromatin(양면성 염색질) 마크를 갖는 유전자들을 예측하기 위해, 약 15,000개의 배아줄기세포(ESC) 전사체와 56개 유전자 좌표에 대해 라벨링된 정보를 사용하여 Geneformer를 fine-tuning했다. 그 결과, 단지 제한된 수의 라벨만으로도 bivalent 상태를 높은 정확도(AUC 0.93)로 예측할 수 있었고, 모델이 학습되지 않은 다른 유전자들에 대해서도 일반화 성능을 보였다. 이는 Geneformer가 전사체 패턴만으로 크로마틴 구조를 간접적으로 추론할 수 있다는 것을 의미한다.
다음으로, 전사인자의 조절 거리(range of regulatory influence)를 예측하는 실험에서는 iPSC에서 심근세포로 분화하는 약 34,000개의 세포 전사체만을 활용해, 특정 전사인자가 단거리 또는 장거리 유전자를 조절하는지를 분류하도록 모델을 fine-tuning했다. 이 역시 ChIP-seq이나 물리적 거리 데이터 없이 수행되었으며, 기존 방법들과 비교해 Geneformer가 월등히 높은 정확도를 보였다. 이로써 Geneformer는 단순한 발현 예측을 넘어, 전사 조절의 복잡한 특성까지 학습하고 추론할 수 있음을 입증했다.

Network dynamics predictions
질병을 유발하는 중심 유전자를 식별하고 이를 조절하는 것은, 단순한 말단 효과 유전자를 교정하는 것보다 치료적으로 더 효과적일 수 있다. 기존에는 이런 중심 유전자를 밝히기 위해, 환자 유래 세포에 다양한 전사체 교란 데이터를 반복적으로 수집해야 했다. 본 연구에서는 Geneformer를 약 3만 개의 정상 심장 내피세포(EC) 전사체로 fine-tuning하여, NOTCH1(N1) 의존 유전자 네트워크 내 중심 유전자와 주변 유전자를 분류할 수 있는지를 테스트했다. 결과적으로 Geneformer는 별도의 교란 데이터 없이도 중심 유전자를 높은 정확도(AUC 0.81)로 예측했으며, N1의 하위 조절 유전자(targets)와 비표적(non-target) 유전자까지 구분할 수 있었다.
또한, 세포 수를 점차 줄여가며 학습시켜본 결과, 단 5,000개의 EC로도 거의 동일한 예측 성능을 유지했으며, 더 나아가 건강한 대동맥과 확장된 대동맥에서 유래한 단 884개의 EC를 사용해도 Geneformer는 중심 유전자를 정확히 예측할 수 있었다. 이 성능은 기존 방법이 3만 개의 일반 EC로 학습한 것보다 더 우수했다. 이는 사전학습(pretraining)의 강력한 효과를 보여주는 결과로, 데이터 양뿐 아니라 해당 데이터가 특정 예측 과제와 얼마나 밀접하게 관련되어 있는지가 학습 효과에 큰 영향을 준다는 것을 시사한다.

Pretraining encoded network hierarchy
Geneformer가 네트워크 동역학을 어떻게 학습했는지를 알아보기 위해, 사전학습된 모델의 attention weight를 분석했다. 각각의 유전자는 학습 과정 중 어떤 유전자에 attention를 기울이는지를 자동으로 학습하는데, 이 weight는 예측 정확도를 높이기 위해 반복적으로 최적화된다. Geneformer는 총 6개의 레이어와 각 레이어마다 4개의 attention head를 가지며, 각 head는 서로 다른 유전자 그룹에 집중함으로써 예측력을 향상시키도록 설계되어 있다.
aortic endothelial cell(EC) 데이터를 분석한 결과, 전체 attention head 중 약 20%는 다른 유전자보다 전사인자(transcription factors)에 더 높은 주의를 기울이고 있었다. 이는 Geneformer가 사전지식 없이도 전사인자가 세포 상태 구분에 중요하다는 사실을 자율적으로 학습했다는 것을 의미한다. 또한, 특정 attention head들은 NOTCH1(N1) 유전자 네트워크 내 중심 유전자(central regulatory nodes)에 주변 유전자보다 더 많은 주의를 기울였다.
흥미롭게도, 모델의 초기 레이어 attention head들은 다양한 유전자 rank를 폭넓게 참조하며 입력을 탐색하는 성향을 보였고, 중간 레이어는 가장 폭넓은 범위의 유전자에 주의를 기울였다. 마지막 레이어에서는 중심 유전자(highly ranked genes)에 집중하는 head가 우세하게 나타났다. 즉, Geneformer는 초기에는 세포 상태를 전반적으로 파악하고, 점차 중심 유전자에 집중하며 정보를 정제해 나가는 방식으로 유전자 간 계층 구조를 학습한 것이다.
In silico gene network analysis
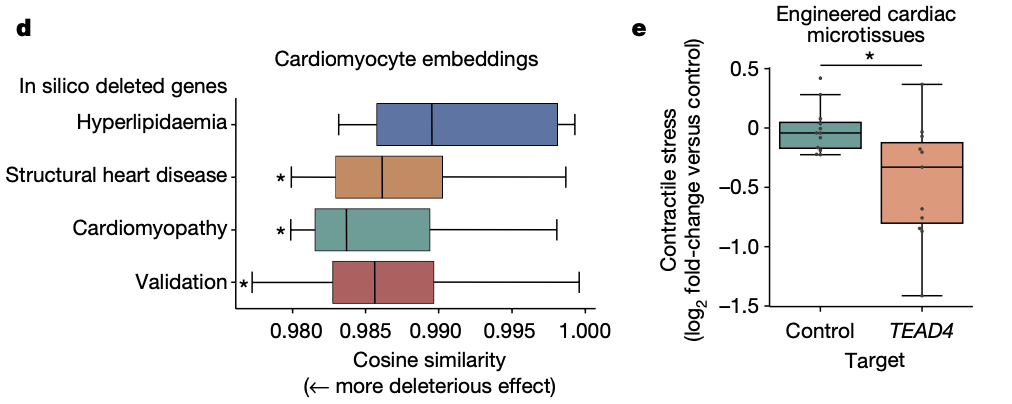
Geneformer의 유전자 임베딩은 네트워크 내 attention weight의 종합적인 결과를 반영하므로, 모델이 사전학습만으로 전사인자와 그 표적 유전자 사이의 관계를 어느 정도 파악했는지 확인하고자 했다. 이를 위해 선천성 심장병과 관련된 전사인자인 GATA4를 fetal cardiomyocyte 세포에서 in silico deletion 하는 실험을 수행했다.
그 결과, GATA4를 제거했을 때 가장 큰 영향을 받은 유전자들은 과거 iPSC 기반 심장병 모델 연구에서 GATA4 변이에 의해 강하게 영향을 받는 것으로 밝혀진 유전자들과 상당 부분 일치했다. 특히, GATA4의 직접 표적 유전자(ChIP-seq 데이터 기준)는 간접 표적이나 housekeeping gene에 비해 임베딩 변화가 더 컸다.(즉, 합리적인 결과)
이와 유사하게 TBX5라는 또 다른 심장병 관련 전사인자에 대해서도 in silico deletion을 했을 때, 역시 직접 표적 유전자들이 간접 표적이나 housekeeping gene보다 훨씬 더 영향을 받았다. 이 결과는 Geneformer가 이미 학습된 임베딩만으로도 유전자 네트워크 구조를 어느 정도 인식하고 있음을 시사한다.
더 흥미로운 점은, GATA4와 TBX5는 상호작용하는 파트너 전사인자인데, 두 유전자를 동시에 in silico deletion 했을 때 그들의 공동 표적(cobound targets)에 대한 영향은 각 유전자를 단독으로 제거했을 때보다 훨씬 더 컸다. 즉, Geneformer는 이 두 전사인자가 협력적으로 작용한다는 생물학적 특성까지 사전학습 기반으로 암묵적으로 인식하고 있었던 것이다.

In silico treatment analysis
연구진은 Geneformer의 in silico perturbation 전략을 확장하여 인간 심장 질환을 모델링하고, 잠재적인 치료 타깃 유전자들을 예측할 수 있는지 실험했다. 먼저, Geneformer를 fine-tuning하여 비정상 심장 상태(비대심근병증, 확장심근병증)와 정상(non-failing) 심장 상태의 심근세포를 구분하도록 훈련시켰고, 이 모델은 테스트 세트에서 90%의 정확도를 기록했다.
이후, 정상 심근세포에서 특정 유전자를 in silico로 삭제하거나 활성화했을 때, 그 세포 임베딩이 비정상 상태(비대심근병증 또는 확장심근병증) 방향으로 얼마나 이동하는지를 측정했다. 그 결과:
- 447개 유전자의 결손이 심근세포를 비대심근병증 상태로 이동시키는 것으로 나타났고, 이 유전자들은 티틴 결합, 근절(sarcomere) 구성과 같은 관련된 경로에 풍부하게 포함되었다.
- 478개 유전자의 결손은 세포를 확장심근병증 상태로 이동시켰고, 여기에 포함된 유전자들은 근수축, 미토콘드리아 기능 관련 경로와 연관되었다.
다음으로, 실제 비대심근병증 또는 확장심근병증 환자의 심근세포에 대해, 특정 유전자를 in silico로 조절(inhibit 또는 activate)했을 때 정상 심장 상태 쪽으로 임베딩이 회복되는지를 분석했다. 이 과정을 통해 치료 후보 경로들이 도출되었고:
- 비대심근병증에서는 ADCY5 (장수 및 심장 보호와 연관된 유전자) 및 SRPK3 (치료 타깃으로 고려 가능한 MEF2 경로 하위 유전자) 등이 유력한 후보로 제시되었다.
마지막으로, 확장심근병증에 대한 Geneformer의 예측을 실험적으로 검증했다. 대표적인 확장심근병증 유발 원인 중 하나인 Titin(TTN) 단백질 절단 돌연변이(TTN+/−)를 보유한 iPSC 유래 심근 조직에서 GSN과 PLN이라는 두 유전자를 CRISPR로 제거한 결과, 이 세포들의 수축력이 크게 향상되었다. 이는 Geneformer가 제안한 유전자들이 실제로 유망한 치료 타깃이 될 수 있음을 입증한 것이다.
Discussion
연구진은 Geneformer라는 문맥 인식형 딥러닝 모델을 개발하여, 대규모 전사체 데이터를 기반으로 사전 학습(pretraining)함으로써 데이터가 부족한 환경에서도 예측을 가능하게 만들었다. Geneformer는 수많은 세포 상태를 관찰하며 학습했고, 이 과정을 통해 유전자 네트워크의 위계와 동역학에 대해 self-supervised하게 되었다.
이 모델은 문맥에 맞는 in silico 유전자 결손 분석을 통해 유전자 복제 수(dosage)에 민감한 질병 유전자를 예측할 수 있으며, 이는 복잡한 형질을 유발하는 GWAS 후보 유전자들의 우선순위를 평가하고, 영향을 미칠 조직까지 예측하는 데 유용하다. 실제로, TEAD4라는 유전자가 태아 심근세포에서 중요한 유전자임을 실험적으로 검증함으로써 Geneformer의 생물학적 통찰력 도출 능력이 입증되었다.
또한, 심근병증과 관련된 질병 모델에서 소수의 환자 샘플만 사용했음에도, Geneformer는 유망한 치료 타깃 후보를 정확히 예측해냈고, 그 후보 유전자들을 조절했을 때 iPSC 기반 질병 모델에서 기능적 개선이 관찰되었다. 따라서 Geneformer는 극히 드물거나 조직 접근이 어려운 질병들에서도, 제한된 데이터로 치료 타깃을 발견할 수 있는 가능성을 보여준다.
아울러, 더 크고 다양한 데이터로 사전 학습을 할수록 Geneformer의 예측력이 향상됨을 확인했으며, 이는 자연어 처리, 컴퓨터 비전 등 다른 분야에서의 관찰 결과와도 일치한다. 다양한 실험 데이터를 사전 학습에 포함한 결과, 단일세포 분석에서 흔히 문제가 되는 배치 간 기술적 오류나 개인 간 변이성에도 강한 robustness을 보였다.
결국, Geneformer는 유전자 네트워크의 근본적인 구조와 작동 원리를 학습한 사전학습 모델로, 연구자들이 제한된 데이터만으로도 의미 있는 생물학적 예측과 발견을 수행할 수 있게 해주는 강력한 도구임을 입증했다.


