제목: Transformers in single-cell omics: a review and new perspectives
출판: Nature Methods (2024년 8월)
요약: single-cell omics 분석에 transformer 모델 활용, 다양한 모델 리뷰 및 한계, 향후 연구방향 제시
Abstract
양질의 데이터셋이 생겨나면서, 현재의 single-cell models의 한계가 두드러졌다.
이에 따라, transformer가 새로운 foundation model로 등장하기 시작하였다. 그 이유는 transformer가 heterogeneous, large-scale datasets의 generalization 능력을 가지고 있기 때문이다.
이 논문에서는 먼저 transformer 구조를 제시하고, single-cell adaptations와 현재 적용 사례를 리뷰하며, 미래의 잠재성에 대해서도 논의하고자 한다.
single-cell omics는 생물학적 시스템을 이해하는데 중요한데, 현재 존재하는 single-cell omics 분석 기법은 대규모 single-cell 데이터셋의 variation을 capturing하는데 한계가 존재한다. 즉, 새로운 computational strategies가 필요하다.
ML분야에서는 NLP task에서 transformer가 처음 등장했지만, 최근에 들어서는 transformer가 다양한 도메인에서 foundation model의 backbone으로 활용되고 있다.
- BERT: Devlin, J., Chang, M.-W., Lee, K. & Toutanova, K. BERT: pre-training of deep bidirectional transformers for language understanding. In Proc. 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies (eds Burstein, J., Doran, C. & Solorio, T.) 4171–4186 (2019).
- Attention: Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst. 30, 5998–6008 (2017).
This work introduced the transformer architecture, originally designed and evaluated on NLP tasks. - Notion of a foundation model: Bommasani, R. et al. On the opportunities and risks of foundation models. Preprint at https://doi.org/10.48550/arXiv.2108.07258 (2021).
foundation model이란, 주로 self-supervision을 통해 대규모 데이터셋에서 학습된 모델로, 다양한 downstream task에 활용될 수 있는 모델이다. transformer-based foundation models 예시는 아래와 같다.
- Avsec, Ž. et al. Effective gene expression prediction from sequence by integrating long-range interactions. Nat. Methods 18, 1196–1203 (2021). => genomics
- Lin, Z. et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science 379, 1123–1130 (2023). => preteomics
- Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024). => preteomics
⭐️위와 같은 도메인의 데이터에서 주목할만한 특성은 sequentiality로, 이 특성은 raw single-cell omics data에서 보이지 않는 특성이므로, 이 부분이 challenging 하다.
⁉️single cell omics data에 시퀀스적인 특성이 없는데 왜 transformer를 활용을 하려고 하는가? ...에 대한 근본적인 의문이 생김 → transformer가 sequence 기반에서 처음 고안된 것은 맞지만, self-attention을 통해 관계를 학습하는 것이 핵심이라 사용했다고 봐도 되는지?

Overview of the transformer architecture
일반적인 딥러닝 모델에서 제일 기본이 되는 개념인 vector. vector는 input의 주된 형태인데, single-cell genomics 분야의 맥락에서는 아래와 같이 표현될 수 있을 것이다. 예를 들면, 각 vector는 각각의 세포를 의미하고, 각 vector의 elements들은 각 유전자의 RNA expression이 될 수 있을 것이다.

가장 대표적인 모델 아키텍쳐는 autoencoder로, 입력을 그대로 복원하는 self-supervised 방식이기 때문에 annotation 없이도 잘 동작한다.
encoder가 input을 받아 low-dimensional latent representation으로 mapping을 하면, decoder가 이를 reconstruct하는 구조.
이 모델이 single-cell RNA counts 데이터에서 학습되면, 세포의 transcriptomic profile을 차원축소(latent representation 생성)할 수 있게 된다.
이 모델은 각 세포 벡터마다 독립적인 파라미터가 학습되는 것이 아니라, 전체 세포에 대해 공통된 고정된 파라미터 집합이 학습된다. 그 과정에서, bottleneck으로 인해 덜중요한 noise는 버리고, 중요한 정보인 모든 세포에서 중요한 특징을 학습하게 된다. 압축시키면서, 나중에 decoder에서 reconstruct했을 때의 오류도 최소화해야하므로, 각 세포의 핵심적인 차이를 구별할 수 있는 표현을 찾게 된다. 즉, cellular variation을 잘 나타내는 latent representation을 얻게 된다.
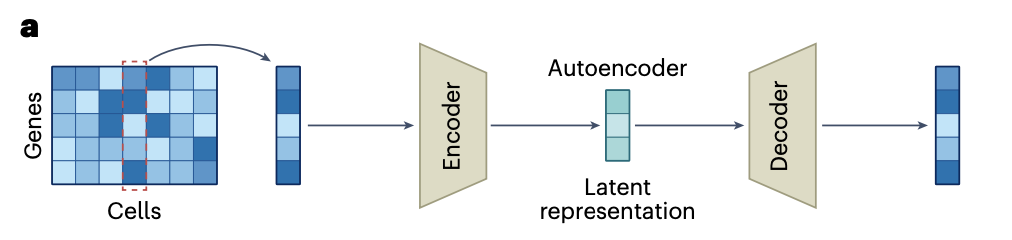
cf. autoencoder의 학습 과정은 mini-batch 학습으로, 하나하나씩 데이터를 학습시키는게 아니라 한번에 n개의 세포 벡터를 모델에 넣고(미니배치) 전체를 여러번 반복하면서 학습하게 된다.
transformer 아키텍쳐
autoencoder와의 유사점: encoder & decoder로 구성, data annotation없이 학습 가능
autoencoder와의 차이점
1) attention mechanism을 사용하기 때문에 샘플에서 어떤 특정한 input feature를 처리할 때, 그것 자체에만 집중하는게 아니라, 다른 input features를 고려하여 처리한다. → 세포 종류에 따라 서로 다른 gene interaction 패턴 포착 가능(ex. 세포A에서는 유전자1,2간의 상호작용이 중요, 세포B에서는 유전자 2,4간의 상호작용이 중요)
2) transformer는 input을 set of embeddings로 받기 때문에, 학습 이전에 해당 포맷으로 변환하는 과정이 필요하다.
ex. 세포 하나는 "gene embeddings의 집합"으로 표현될 수 있다. (아래 그림 참고)
3) encoder, decoder의 기능, 디자인이 autoencoder에서와 다르다. autoencoder에서 encoder는 input→latent representation을 수행하여 차원 축소를 시켜주는 역할이었다면, transformer에서 encoder를 통해서는 차원축소가 되지 않고, contextualized gene embeddings를 생성하게 된다. (bottleneck의 제약을 받지 않으므로 유전자 간의 관계를 풍부하게 반영 가능)
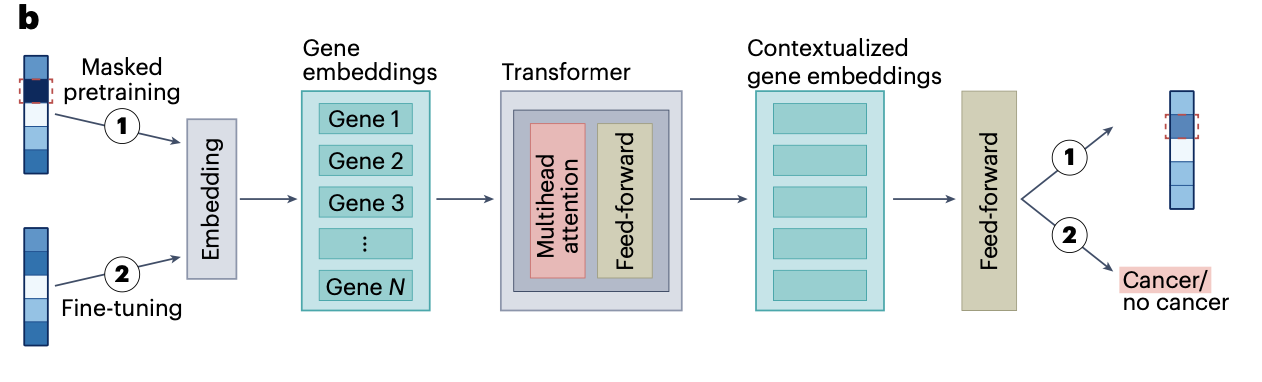
transformer decoder는, encoder 부분의 output을 프로세싱하고, 한번에 한 element씩 sequence 생성한다.
encoder, decoder는 독립적으로 쓰이기도 해서, encoder-only models, decoder-only models가 존재한다.
encoder-only models는 유전자별로, contextualized input embeddings를 생성할 때 사용된다. 여기서 해당 임베딩은 특정 유전자의 특성, 해당 세포에서의 발현량, 다른 유전자들의 발현정보를 함께 반영한 것이기 때문에 gene-level tasks(ex. gene dosage sensitivity prediction) 를 수행할 수 있다.
Decoder-only models와 encoder-decoder transformers는 sequence생성(ex. transcriptomic cell profile simulation)에 활용가능한데, 이때 입출력 모두 시퀀스 형태여야 해서 RNA 발현량 데이터를 시퀀스 형태로 표현해야 한다. [유전자1발현량 유전자2발현량 유전자3발현량 ......]
Representing single-cell input data
앞서 언급했듯이, single-cell omics data는 nonsequential 하기 때문에 transformer에 넣기 위해서는 적합한 포맷으로 데이터를 임베딩하는 과정이 필요하다.(challenging한 부분)
input sample을 집합 S, 해당 집합을 구성하는 임베딩을 Xi 라고 하면, S: 한 조직 내 세포들의 집합, Xi: 각 세포를 의미할 수도 있고, S: 한 세포 내 유전자들의 집합, Xi: 각 유전자들의 특징(ex.RNA발현량)을 의미할 수도 있다.
Transformer는 input embeddings의 순서가 중요하지 않기 때문에, positional encoding이 등장하게 되었다.
⁉️그렇다면 single-cell omics data를 활용할 때는 해당 positional encoding을 제외하면 되는거 아닌가?
single-cell omics data를 적합한 transformer inputs로 변환하는 접근 방식은 총 3가지 정도가 존재한다.(fig.3)
input sample S는 transcriptional profile of a cell를 담고 있다고 가정
transcriptional profile of a cell: 세포 안에서 어떤 유전자들이 얼마나 발현되고 있는지를 보여주는 정보
1. Ordering
데이터는 자연어 처리(NLP)에서처럼 토큰들의 시퀀스(순서 있는 나열)로 표현되며,
이는 NLP 트랜스포머 모델을 그대로 재사용할 수 있게 해준다.
iSEEEK, Geneformer, tGPT: 각 유전자가 토큰에 해당, 유전자 토큰의 순서는 해당 세포 내에서 normalized expression 기준으로 정렬하여 결정된다.(fig3.c)
⁉️발현량의 값 자체가 반영되고 그 순서를 정하는데 ranking이 사용된 것이 아니라, 발현량은 무시되고 ranking값만 반영..??? → gene embedding에다가, 발현량을 기준으로 ranking 매긴대로 positional encoding 값 부여했기 때문에 발현량 값이 직접적으로 반영된 것은 X
이 방식은 단일세포 데이터를 시퀀스로 바꾸는 데 도움이 되지만, 데이터 해상도(data resolution)를 낮추고, 정보 손실(information loss)이 발생할 수 있다.
2. Value categorization
각 유전자의 RNA counts가 value binning을 거침(fig.3d)
continuous -> categorical data
gene embedding + value bin embedding
single-cell transformer scBERT: bin의 간격이 동일; 어떤 경우에는 대부분의 발현값이 단 하나의 구간에 몰리는 현상을 보였고,
이로 인해 데이터의 해상도가 심각하게 저하되는 문제가 발생할 수 있다.
scGPT: bin의 간격이 adaptively sized; 예를 들어 한 세포에서 1,000개의 유전자가 발현되었다면 이를 발현량 순으로 정렬한 뒤, 100개씩 끊어서 10개의 구간(bin)으로 나눔; 가장 높은 발현량은 가장 높은 bin에 해당할 것이므로, semantic meaning도 유지
3. Value projection
또 다른 접근법으로는 입력 데이터를 projection하는 방법이 있다.(fig.3e)
예를 들어, 입력 데이터가 한 세포의 transcriptomic profile—즉, 유전자 발현값의 벡터—로 주어진 경우,
각 입력 xi는 두 가지 요소의 합으로 구성된다:
- 유전자 발현 벡터에 대한 projection
- 위치 정보나 유전자 임베딩(positional or gene embedding)
이 방식은 1,2 방식과 달리 데이터 해상도가 반드시 떨어지지는 않는다. continuous value 자체를 그대로 임베딩에 사용하므로, 기존 categorical token 기반 임베딩을 사용하는 NLP transformers들과는 다르다. (성능에 미치는 영향은 아직 명확X)
트랜스포머에 입력되는 임베딩의 개수는, 반드시 입력 데이터에 포함된 유전자 수와 일치할 필요는 없다.
예를 들어, 출력 임베딩의 수는 선택된 유전자 경로(gene pathways)의 수와 일치할 수도 있다.
실제로 TOSICA 모델에서는 이렇게 설계함으로써 모델 해석 가능성(interpretability)을 높였다.
주의) gene embedding != RNA expression value
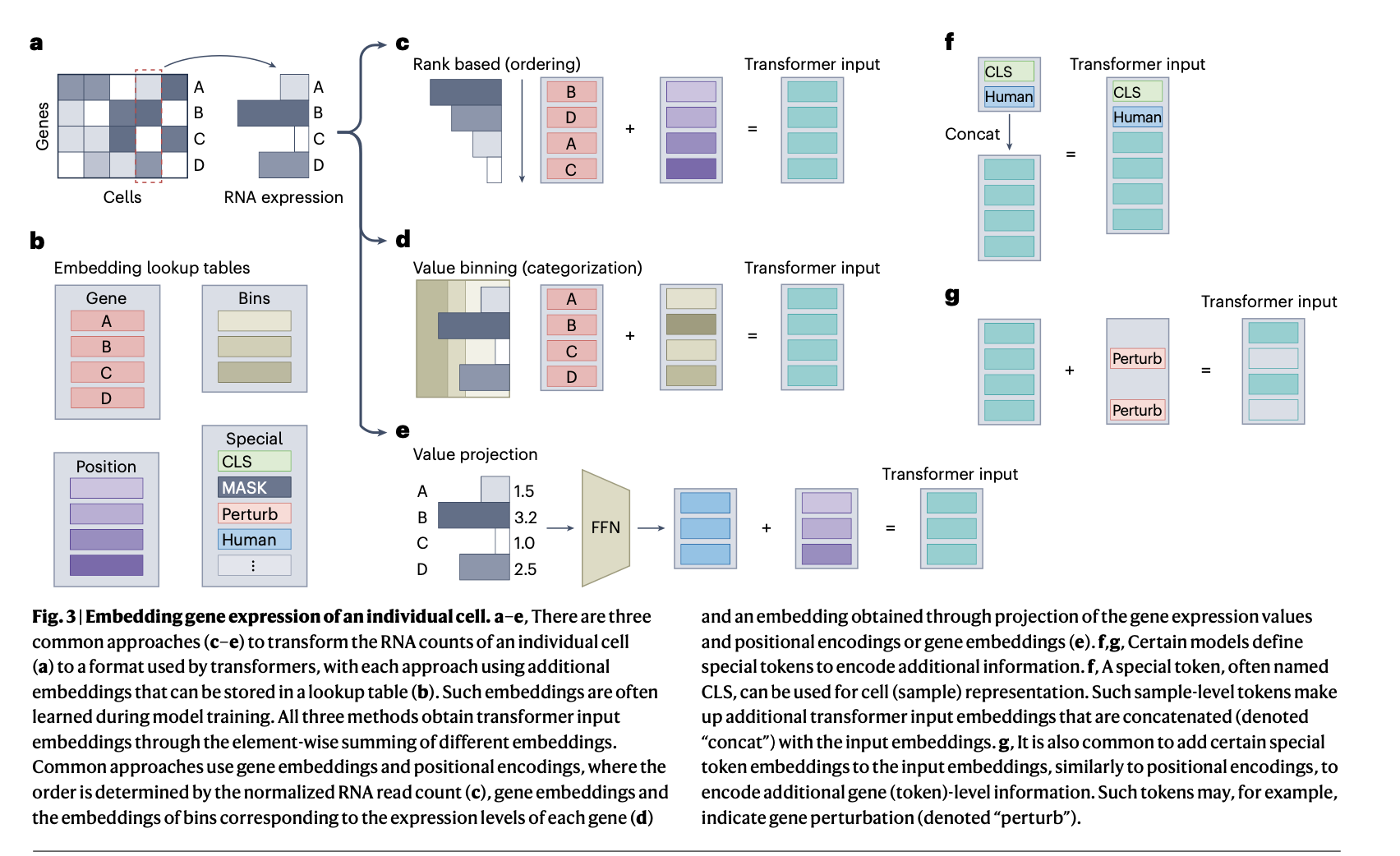
또한 트랜스포머는 special tokens을 활용함으로써, 입력에 extra layers of information을 포함시킬 수 있다.
이러한 speical tokens은 예를 들어, pertubation, species 정보, batch 정보, 데이터모달리티와 같은 정보를 나타낼 수 있다.
이 special token embedding은 다음 두 가지 방식 중 하나로 모델에 들어간다:
- 토큰 수준(token-level)의 정보일 경우
→ 각 입력 벡터 xi에 더해짐(concat) (Fig. 3f) - 샘플 수준(sample-level)의 정보일 경우
→ 전체 입력 세트 S에 새로운 토큰으로 추가됨 (Fig. 3g)
이러한 특수 토큰 임베딩들도 보통 학습 가능한 벡터이며, 유전자 임베딩이나 위치 임베딩처럼 훈련 과정에서 업데이트된다.
만약 S가 하나의 조직 안에 있는 여러 세포들의 집합이고, Xi가 각각의 개별 세포를 나타낸다면, Xi는 positional encoding+expression profile embedding으로 모델링될 수 있는데, 이때 positional encoding은 세포의 공간좌표로부터 계산된다.
expression profile embedding은 Σ (gene_embedding × gene_expression_level)로 계산할 수 있다. 이러한 방식은 cell-cell relationship을 모델링하는 데에는 유리하지만, gene-gene interaction 분석은 제한적이다.
Gene and cell attention
transformer layer는 attention layer + feed-forward network(FFN)으로 구성
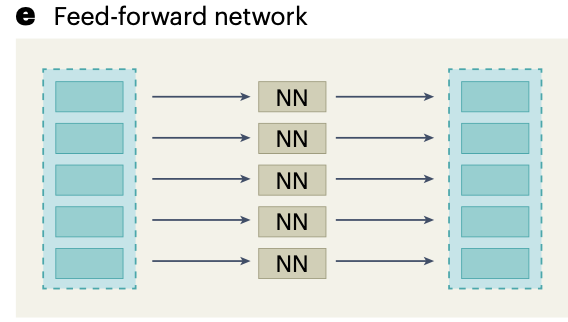
single-cell omics에서 input embeddings는 주로 유전자에 대응되므로, 유전자에 대해 attention을 적용하는 것은 곧 유전자간의 관계 패턴을 포착, gene regulatory network(GRN)을 구축할 수 있다는 것을 의미한다.
multihead attention을 활용하면 아래와 같이 DNA repair에서의 gene-gene relationships, apoptosis에서의 gene-gene relationships 등등..을 포착할 수 있게 된다.

gene-gene interaction뿐만 아니라, (17,20-22,24)
- Theodoris, C. V. et al. Transfer learning enables predictions in network biology. Nature 618, 616–624 (2023).
This work proposed the first single-cell transformer that has successfully predicted candidate therapeutic targets. - Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods https://doi.org/10.1038/s41592-024-02201-0 (2024).
This work proposed a single-cell transformer architecture that has been used for a wide range of tasks, including perturbation response prediction and multiomic data integration - Shen, H. et al. A universal approach for integrating super large-scale single-cell transcriptomes by exploring gene rankings. Brief. Bioinform. 23, bbab573 (2022). This work introduced the first gene-ranking-based single-cell transformer. It was also the first single-cell transformer pretrained on a large dataset of over 10 million cells.
- Shen, H. et al. Generative pretraining from large-scale transcriptomes for single-cell deciphering. iScience 26, 106536 (2023).
- Yang, F. et al. scBERT as a large-scale pretrained deep language model for cell type annotation of single-cell RNA-seq data. Nat. Mach. Intell. 4, 852–866 (2022).
cell-cell interaction을 모델링하는 방식도 제안되었다. (이 방식에서는 공간좌표 정보를 positional encoding에 주로 넣음)
- Wen, H. et al. CellPLM: pre-training of cell language model beyond single cells. In International Conference on Learning Representations (eds Kim, B. et al.) (2024).
- Wen, H. et al. Single cells are spatial tokens: transformers for spatial transcriptomic data imputation. Preprint at https://doi.org/10.48550/arXiv.2302.03038 (2023).
하지만 이 논문에서는 single-cell focus로, gene-centric attention에 대해 리뷰하고 있다.
아래 fig.2c,d에서는 차례대로 multihead attention, attention mechanism을 보여주고 있다. multihead attention의 경우 attention head를 concat하는 것. attention mechanism에서는 각 weight를 곱해 query, key, value를 얻고, query와 key간의 유사성 계산을 통해 attention score을 얻을 수 있다. 정규화한 attention score은 value vector의 weighing factor로 작용한다. 즉, value vector에 곱해서 결과를 얻는다.
attention 계산 과정에서, O(n^2)의 복잡도가 생기므로, 만약 집합 S의 input element개수가 많다면 문제가 될 수 있다.
이 문제를 해결하기 위해, FlashAttention, Performer, sparse attention, iterative attention이 제안되었다.
- FlashAttention: Dao, T., Fu, D. Y., Ermon, S., Rudra, A. & Ré, C. FlashAttention: fast and memory-efficient exact attention with IO-awareness. Adv. Neural Inf. Process. Syst. 35, 16344–16359 (2024).
- Performer: Choromanski, K. M. et al. Rethinking attention with performers. In Proc. 9th International Conference on Learning Representations (eds Hofmann, K. et al.) (2021).
- sparse attention: Roy, A., Saffar, M., Vaswani, A. & Grangier, D. Efficient content-based sparse attention with routing transformers. Trans. Assoc. Comput. Linguist. 9, 53–68 (2021).
- iterative attention: Jaegle, A. et al. Perceiver: general perception with iterative attention. In Proc. 38th International Conference on Machine Learning (eds Balcan, N. et al.) 4651–4664 (2021).


효율적인 attention 계산 및 attention 근사 기법, 그리고 DeepSpeed와 같은 일반적인 딥러닝 가속 프레임워크는 여전히 활발히 연구되고 개발되고 있는 분야이며, 향후 모델들이 모델 용량(모델 파라미터 수)과 입력 차원수(예: 유전자 수) 양쪽 측면에서 확장될 수 있도록 해줄 것으로 예상된다.
최근에는 다음과 같은 최적화 기술들이 다양한 single-cell transformer에 이미 적용되고 있다:
- DeepSpeed (Geneformer에서 사용)
- FlashAttention (scGPT과 Cell2Sentence에서 사용)
- Performer (scBERT에서 사용)
attention scores는 multiple layers, attention heads에서 summation 등이 일어나는데, 이는 해석력을 떨어뜨린다. 어텐션 스코어와 실제 피처의 중요도(feature importance)는 항상 일치하지 않으며, 따라서 어떤 입력 요소가 최종 출력에 영향을 주었는지를 알아내기 위한 신뢰할 수 있는 도구로 간주되기는 어렵다.
⁉️multihead attention 질문: 각각의 attention head에서 attention score을 계산하고 그것도 concat을 하는건지 아니면 sum, avg? sum을 한다면 정보손실이 일어날 것 같은데... 각각의 head에서 목적으로 하는 task(DNA repair head, cell cycle head등..)이 다른데 이거를 합하면 정보손실이 일어날 것 같은데 다른 방식은 없을까?
→ 다시 원래 차원대로 되돌려야하기 때문에 multihead attention을 거치고 Wo를 곱함. 그러면 이 Wo가 학습되면서 각각의 attention 결과를 잘 합치도록 반영되는것인지?
Encoder and decoder
- encoder: process the input data and generates latent representations
- decoder: outputs a sequence based on these representations
encoder, decoder는 약간의 다른 아키텍쳐를 가지고 있지만, 둘다 mutiple layers of attention + FFNs
decoder는 self-attention을 활용하는 encoder와 다르게, masked attention, encoder-decoder attention(cross attention)을 활용한다.

1. 지금까지, 대부분의 single-cell transformers는 encoder-only 아키텍쳐에 의존하였다. 다음 섹션에서 설명하겠지만, 이는 효율적인 pretraining strategy인 masked language modeling(MLM)을 가능하게 한다.
2. decoder-only model를 이용하는 tGPT, scMulan도 뛰어난 성능을 보였다. 이러한 모델은 조건부 데이터 생성을 가능하게 하는데, 예를 들어 세포 종류, 기증자 나이, 시퀀싱 기술 등이 주어졌을 때 그에 대응하는 transcriptomic profile을 생성할 수 있다.
3. 일부 모델들은 트랜스포머 레이어를 인코더 또는 디코더 구조를 기반으로 한 variants으로 사용한다.
예를 들어, scGPT는 디코더의 마스킹 어텐션에서 영감을 받은 맞춤형 masked attention을 사용하여, autoregressive generation을 가능하게 한다.
Training transformers
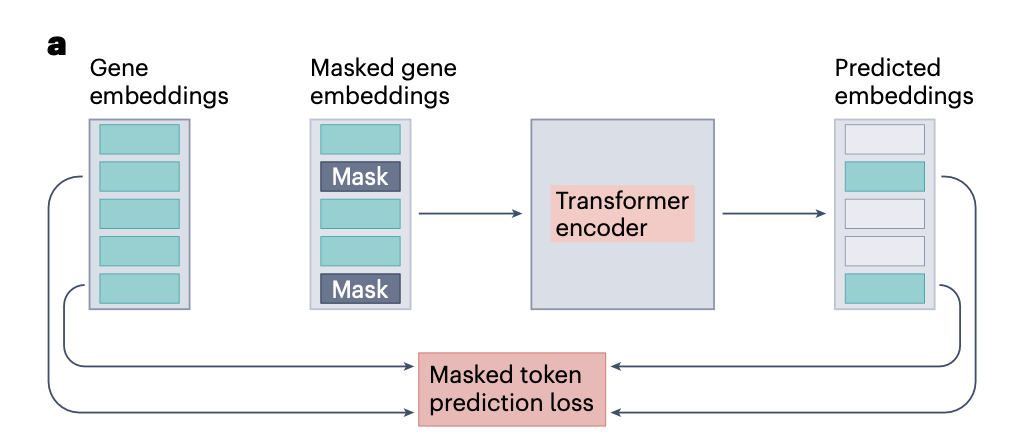
cell annotation의 한계, 부족 → masked-token, next-token prediction(NTP)로 구현된 self-supervised learning(SSL) 활용
대표적인 self-supervised pretraining 예시는 MLM.
MLM은 특정 input tokens를 MASK token으로 대체하고, 해당 부분을 예측하게 만드는 방법이다.
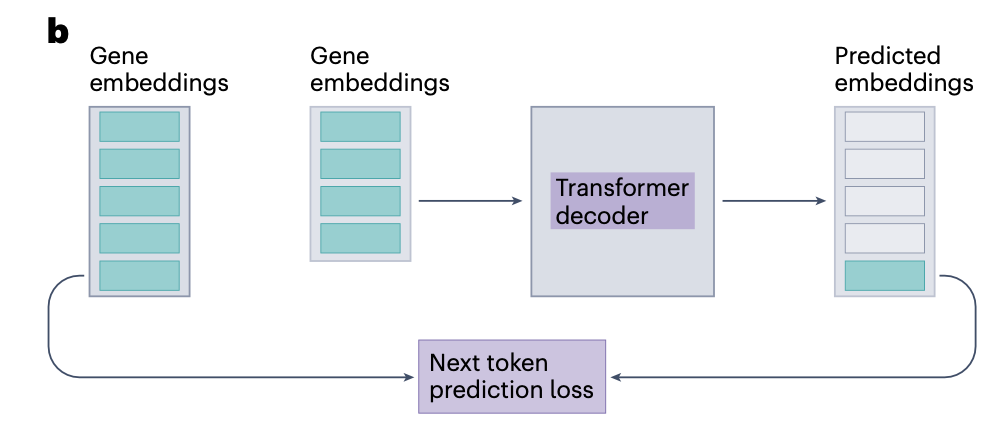
대안으로 transformer decoder, encoder-decoder model에서 NTP task 방법이 쓰이기도 한다. 이는 입력이 시퀀스라고 가정하고, 다음 토큰을 예측하도록 모델을 학습시키는 방식이다.
⇒ 무조건적으로 SSL(self-supervised learning)을 활용하는 것이 좋은 건 아니고, 만약 annotation이 충분히 존재한다면 그냥 supervised learning을 하는게 결과는 더 좋게 나올 것 같다. 상황에 따라 판단하기!
Applications of single-cell transformers
cf. CLS : 입력 전체를 대표하는 임베딩을 만들기 위해 붙이는 추가 토큰; 세포 전체의 요약 표현

cell type annotation, cross-modality prediction, gene representation learning 등 여러 task에 transformer를 활용할 수 있다.
희망사항: 대용량의 데이터에 모델을 학습시켜서, 세포생물학에 대한 foundational knowledge를 습득하도록 함 → 다양한 downstream task에 해당 foundation model을 fine-tuning 등을 거쳐 활용하기
Challenges in modeling single-cell data
- Noise: 기술적인 요인으로 인한 노이즈; 해결방안: 하나의 데이터셋보단 여러 데이터셋을 본 모델이 노이즈에 더 robust할 것, value categorization 활용해서 신호 퀄리티 향상하기도함
- Batch effects: 데이터가 생성된 환경이 다름으로 인해 발생; 해결방안: batch tokens 활용 또는 heterogeneous datasets에 모델을 노출시켜서 배치효과 완화
- Sparsity: large fraction of value zero... 해결방안: processing only nonzero values, grouping features into pathways
Gene representation
유전자 발현을 input embeddings로 인코딩했다는 것은, 그 과정에서 유전자간의 attention을 활용해 인코딩된 gene embeddings는 context specificity를 가지게 된다. 이러한 contextualized gene embeddings를 활용해 기능적으로 비슷한 유전자를 찾거나, 유전자 기능 예측 등의 task를 수행할 수 있다.
예시1) pretrained scGPT에서 gene embeddings를 통해 기능적으로 관련있는 유전자 similarity networks 구축
예시2) Geneformer의 gene embeddings를 fine-tuning해서 chromatin states 예측, 특정 세포 종류의 유전자 네트워크에서 중심, 주변부 요소 구분 등에 활용
예시3) transformer 모델에서 생성된 embedding이 다른 모델에 정보제공자처럼 활용 가능: GEARS(pertubation response prediction model)와 같이 사전정의된 유전자 임베딩을 사용하는 모델에 transformer에서 생성한 임베딩을 입력값처럼 사용 가능
예시4) 종간 분석에서 transformer를 사용하면 context-specific orthology mapping 가능
Interaction between omic features
omics: genomics, epigenomics, transcriptomics, preteomics, metabolomics ....
이러한 omic feature 간의 interaction은 매우 중요하다.
transformer는 이러한 omic features간 attention mechanism을 통해 multimodal interaction을 포착하게 해준다. 한 예시로, 세포와 omic feature token 간의 attention score은 cell type marker genes, hub genes를 찾는데 활용할 수 있다.
- Geneformer의 gene attention scores → reveal cellular regulatory mechanisms
- TOSICA의 pathway attention scores → capture cellular trajectories, link changes
- scGPT의 gene attention scores → GRNs 추론, genetic pertubations이 네트워크에 미치는 영향 분석
하지만, attention score가 절대적으로 해석가능성에 도움이 되는 것은 아니다. transformer의 attention scores는 일부 경우에 feature importatnce와 상관없다는 연구도 있기 때문에, attention scores를 일반화하여 신뢰하는데 주의가 필요하다.
- Serrano, S. & Smith, N. A. Is attention interpretable? In Proc. 57th Annual Meeting of the Association for Computational Linguistics (eds Màrquez, L. et al.) 2931–2951 (2019). 이 논문 참고!
Cell representation
저차원 공간에서 high-quality로 각각의 세포를 표현할 때에는, cell type, state같은 biological variations는 잘 보존하면서, batch effect와 같은 기술적 요인들은 최소화하는 것이 중요하다. 이전까지는 이런 것을 최소화하는 것이 어려웠는데, transformer는 배치 정보를 고려하지 않는 사전 학습(batch-unaware pretraining)을 통해 이 문제에 대한 해결책을 제시한다.
예를 들어, Universal Cell Embeddings (UCE)와 GeneCompass는 연구, 조직, 종을 넘나들며 대규모 분자 프로파일을 통합하는 데 사용되었으며, UCE는 모델이 본 적 없는 종의 데이터에도 cell type annotations을 전달할 수 있었다.
| Variational autoencoders(VAEs): scVI, scArches, SCimilarity | Transformer |
| 명시적으로 저차원 임베딩 학습 | 저차원 세포 임베딩을 직접 생성X, 세포 임베딩은 개별 세포에 대한 transformer 출력 임베딩을 pooling함으로써 얻거나 CLS 토큰 추가.. |
| batch covariates와 input feature(ex.세포의 유전자 발현)를 함께 전달 | batch covariates를 명시적으로 사용하지 않아도 robust |
| 유연성 O: multimodal features 활용 용이 |
트랜스포머 세포 임베딩의 또 다른 활용 예로는 perturbation에 따른 세포 임베딩 변화 분석을 통해 치료 표적을 식별하는 방법이 있다. 예를 들어, Geneformer에서는 비대성 또는 확장성 심근병증 환자의 심근세포(cardiomyocytes) 임베딩이 in silico gene deletion 후 정상 심장 상태에 대응하는 임베딩 쪽으로 이동하는 것을 관찰함으로써, 해당 유전자의 억제가 심근세포 기능 개선에 도움을 줄 수 있음을 나타냈다. 이 예측 결과는 실험적으로 검증되어, 단일 세포 트랜스포머의 유용성을 입증했다.
Single-cell modality generation
single-cell modality generation: 개별 세포에서 유전자 발현 예측, 혹은 다른 omics modality 예측
예시1) scGPT는 Perturb-seq 데이터셋의 일부 유전적 교란(subset of genetic perturbations)을 대상으로 fine-tuning되었으며, 어떤 유전자가 교란되었는지 표시하는 특수 토큰을 사용했다. 이 모델은 이전에 보지 못한 교란에 대해서도 유전자 발현 반응을 정확히 예측함.
예시2) generative transformers는 omic feature 없이도 단지 조건만 입력으로 받아 데이터를 직접 시뮬레이션할 가능성도 있다. 이러한 모델이 개발되면, 침습적 조직 검체 채취나 1상 임상시험과 같이 대조군 조직 획득이 어려운 상황에서 교란 모델링 및 대조군 데이터 예측에 활용될 수 있을 것이다.
Cell annotation
Cell annotation의 주된 focus는 cell type prediction. scBERT, TOSICA와 같은 모델들은 이 작업에만 집중한다.
대규모 데이터셋을 대상으로 masked input modeling을 통한 self-supervised learning 또한 classification 능력을 향상시키는 것으로 알려져있다.
Transformer는 이전에 보지 못한 데이터셋에도 일반화할 수 있는 것을 보여주었는데, 이는 특히 reference dataset을 활용해 새로운 데이터셋에 annotation을 할 때 중요하다. (TOSICA, UCE 모델)
그럼에도 불구하고, non-deep learning 세포 유형 예측기를 능가하는 것은 여전히 어려운 경우가 많다. 예를 들어, 기관별(organspecific) 세포 유형 주석에서는 로지스틱 회귀(logistic regression)가 더 복잡한 비선형 예측기들과 대등한 성능을 보이는 경우가 자주 있다. 기관 간(cross-organ) 세포 유형 주석에서는 트랜스포머가 아닌 모델인 scTab가 비딥러닝 최첨단 세포 유형 예측기와 scGPT를 모두 능가하는 것으로 보고되었다.
또한, 세포 유형 주석이 풍부한 데이터는 세포 유형 주석 작업의 평가를 용이하게 하지만, 데이터셋 간 주석이 서로 일치하지 않는 경우가 많으며 주석의 신뢰성도 문제시되고 있다. 따라서 cell type annotation 작업이 모델 성능을 신뢰할 수 있는 지표라고 보기는 어렵다.
Spatial omics
이 섹션에서는 슬라이드 이미지(h&e와 같은)가 아니라, 수치 기반의 spatial omics 데이터만 다룸.
- Nicheformer: 공간 내 이웃 세포 밀도 예측, 이웃 세포 구성 예측; 공간분석과 RNA-seq 분석 간의 transfer learning 가능하게 함→분리된 세포의 유전자 발현만으로도 공간적 위치(context) 예측할 수 있게 해줌
- scGPT, SpaFormer, CellPLM: ST 데이터에서 유전자 발현 보간(imputation) 작업에 효과적; SpaFormer와 CellPLM은 입력에 세포의 공간좌표를 positional encoding으로 변환하여 사용
공간 전사체학(spatial transcriptomics) 분야는 빠르게 성장하고 있으며, transformer는 다른 분야에서 공간 좌표를 잘 처리해온 경험이 있어, 이 두 기술의 융합은 매우 유망한 연구 방향으로 주목받고 있다.
⁉️흥미가 가는군....
LLMs for single-cell analysis
자연어에 대해 학습된 트랜스포머, 즉 대형 언어 모델(LLMs)을 이용하여 단일 세포 데이터를 모델링하려는 시도들도 이루어지고 있다.
일부 방법들은 자연어 모델(language model)을 직접 사용하는 방식이며, 또 다른 방법들은 molecular data를 텍스트 형식으로 변환한 후 이를 fine-tuning한다.
GPT-4는 일반적인 언어 데이터셋으로 학습되었지만, cell type annotation 및 다양한 바이오메디컬 작업에서 특별한 과제별 학습 없이도 가능성을 보여준다. 또한, 범용 LLM(Generalist LLM)에서 생성된 임베딩은 단일 세포 작업, 예를 들어 batch integration, 세포 유형 분류, 유전자 특성 예측(gene property prediction) 등에 유용한 것으로 나타났으며, 이는 LLM이 single cell에 대한 지식을 어느 정도 내포하고 있음을 시사한다.
초기 시도들에서는, single-cell omics data를 텍스트로 변환한 뒤, 사전 학습된 LLM에 fine-tuning하여 그 성능을 활용하려 했다.
예를 들어, Cell2Sentence와 같은 모델은 세포의 전사체 데이터를 유전자 발현량 순으로 나열한 시퀀스로 표현하여 텍스트처럼 입력하고, 여기에 추가적인 설명적 텍스트(descriptors)도 결합할 수 있도록 한다. 이는 기존의 유전자 발현 순위 기반 임베딩 방식과 유사하지만, 텍스트 기반 모델의 장점을 더할 수 있다는 점이 다르다.
또한, 일부 특화된 멀티모달 트랜스포머 모델은 일반적인 텍스트에 대해 사전 학습된 후, 의학 문서, 이미지, 유전체 데이터에 대해 추가로 학습을 진행해, 여러 바이오메디컬 작업에서 범용 모델보다 뛰어난 성능을 보이기도 했다. 이러한 결과는 범용 언어 모델을 기반으로 특화된 단일 세포 모델을 구축하는 것의 가능성을 보여준다.
하지만 여전히, 자연어와는 근본적으로 다른 구조를 가지는 단일 세포 오믹스 데이터와 텍스트 중심 모델을 효과적으로 융합하는 문제는 미해결 과제로 남아 있다.
Foundation models
Foundation model은 unannotated large dataset에서 학습하는 범용 AI 모델로, 다양한 downstream task에 적은 데이터만으로도 적용이 가능하다.
- 대표 예시:
- NLP: BERT, GPT-4
- 음성: Whisper
- 이미지: Vision Transformer
Transformer는 entities간 관계를 모를 때에 유리하고, 유연성, 확장성 덕분에 GNN보다 널리 쓰이고 있다. GNN과 transformer의 장점을 결합하는 모델도 개발중에 있다.
Single-cell foundation model은 대용량의 데이터에서 훈련된 ML model로, 다양한 방면으로 활용될 수 있다.
- 대표 예시:
- SCimilarity: autoencoder 기반
- GeneCompass (12-layer transformer, 1억 개 파라미터, 1.2억 개 세포 학습)
- UCE (현재 파라미터 수 기준 가장 큼: 33 layers, 6.5억 개 파라미터, 3천6백만 세포 학습)
Single-cell foundation model은 대용량의 데이터에서 훈련된 ML model로, 다양한 방면으로 활용될 수 있다.
이러한 거대한 모델들이 생겼지만, 아직은 간단한 task-specific 모델들이 더 잘 작동하는 경우가 많으며, 비용, 자원이 훨씬 덜 드는 모델이 오히려 성능이 좋을 때도 있다.
⁉️머신러닝에서 데이터가 단순하면 모델이 복잡한것보다 오히려 단순한게 더 잘 작동하는데.. 혹시 이런거랑 같은 원리? 혹시 이 경우도 single-cell 데이터가 단순한 편이어서 오히려 간단한 모델들이 더 잘 작동하는건지... 아니면 그냥 거대 모델들의 잠재성이 더 크긴하지만 아직 연구가 많이 안되어서 그런건지 ??
Current limitations and evaluation of models capabilities
1. Single-cell 데이터에 sequence 구조가 명확하지 않고, rank ordering, chromosome 위치 기반 정렬은 임시방편 수준이다.
2. 엄밀한 벤치마크가 필요하다. 벤치마크가 있어야 어떤 것이 optimal data encoding, architecture, training procedure인지 알 수 있게 될 것이다. 따라서 몇 가지 transformer의 벤치마크가 수행되었는데(참고문헌 36, 41, 77-79), 여전히 단순한 모델이나 task-specific model이 transformer의 성능을 능가하고 있으며, transformer의 zero-shot performance가 약하다고 한다.
예를 들어, SCimilarity는 autoencoder 기반인데도, 다양한 single-cell datasets을 통합하는 잠재성을 보인다.
⇒ transformer는 single-cell 분야에서 아직 엄청 유의미하다!라고 판단할 수 없음..
벤치마크에도 여러 문제점이 존재하는데, 아래와 같다.
| ① 불완전한 비교 | 경쟁 모델의 하이퍼파라미터 튜닝 누락, 강력한 기존 모델(XGBoost, scVI 등) 무시 |
| ② 생물학적 task의 표준 평가 지표 부족 | perturbation 효과 예측 등은 universally accepted metric이 없음 |
| ③ 대규모, 다양한 데이터에 대한 평가 부족 | 모델 성능 평가에 대한 문제 존재 가능 |
| ④ 데이터 누수(leakage) | pretraining 때 본 데이터가 evaluation set에 포함될 가능성 있음 |
| ⑤ 벤치마킹 자체가 복잡 | 훈련 코드, 데이터셋 접근 어려움 + compute 자원 부담 큼 |
오믹스 데이터를 직접 다루는 모델들과는 별도로, 생성형 언어 모델(generative language models, LLMs)을 단일세포 task에 평가할 때는 고유한 어려움이 있다. 이 모델들은 텍스트 생성을 목표로 학습되었기 때문에, 그 출력이 원하는 형식에 맞지 않을 수 있다.
게다가, 많은 최첨단 LLM들은 공개되어 있지 않으며, 인터페이스를 통해 얻은 결과도 시간에 따라 변할 수 있다.
따라서, LLM을 단일세포 과제에 평가할 때는 특별한 평가 방법이 필요하다.
현재 단일세포 트랜스포머에 대한 평가 방식에는 불일치(discrepancy)가 많아 혼란을 초래하고 있으며, 다른 분야처럼 체계적인 평가가 이 분야에도 도움이 될 것이다. 또한, 기여가 열려 있는 투명한 공개 리더보드는 모델 간 비교의 신뢰도를 높이는 데 기여할 수 있다.
Conclusion and outlook
단일세포 데이터의 양이 계속 증가함에도 불구하고, 다양한 조직, 조건, 종에 걸쳐 세포를 함께 모델링하는 문제는 아직 해결되지 않았다.
현재까지의 주요 전략은, 관심 있는 특정 데이터셋(주로 단일 장기와 단일 종에 한정됨)에 대해 저차원(batch-corrected) 세포 임베딩(cell embeddings)을 생성한 후, 학습된 표현을 활용해 후속 분석을 수행하는 방식이다.
single-cell transformers는 이에 대한 대안적인 접근을 제공할 수 있다.
omics data의 공동 모델링은, 각각의 오믹스 모달리티가 제공하는 다양한 관점을 통합하여 세포 생물학을 정확히 인코딩하는 데 도움이 될 수 있다.
현재의 단일세포 트랜스포머들은 여러 모달리티를 통합하긴 하지만, scRNA-seq 외의 관측치(readout)를 포함하는 연구는 극히 일부만 포함하고 있다. 따라서, 대규모 다중오믹스 트랜스포머의 개발은 여전히 미래 연구를 위한 목표로 남아 있다.
하지만, 도전과제도 포함하고 있는데, 더 많은 모달리티를 포함하게 되면, 일반적으로 더 긴 입력 시퀀스가 필요하게 되기 때문이다.
이러한 메모리 및 계산량 부담을 줄이기 위해, 모델은 예를 들어 0이 아닌 값만 고려하는 희소 표현 방식(sparse representation),혹은 원시 오믹스 특징을 pathway 단위의 의미 있는 묶음으로 그룹화하는 방식 등을 사용할 수 있다.
기계학습 전반에서는 지금도 다음과 같은 질문이 계속 논의되고 있다:
“미래에는 모든 도메인과 사용 사례에 적용 가능한 하나의 다중모달 모델이 등장할까?”
이러한 범용 기반 모델(universal foundation model)은, 텍스트, 이미지, 오디오 같은 다양한 모달리티를 넘나들며,
자연어 인터페이스를 통해 일반 대화, 의학적 진단, 과학 실험 등과 같은 모든 예시들을 연결하는 것을 목표로 한다.
반대로, 도메인별 또는 모달리티별로 설계된 기반 모델(foundation model)을 그 도메인에 특화된 방대한 데이터로 훈련시키는 방안도 있다. 최근의 multimodal biomedical foundation model 제안들은 후자의 방식을 따르고 있으며,
그러한 예시들은 단일세포 유전체학뿐만 아니라 다양한 분야에서도 나타나고 있다.
이와 더불어, 세포 프로파일의 텍스트 표현(textual representations)에 대한 연구도 병행되고 있으며, 이는 생물학, 의학, 화학을 포함한 다양한 과학 분야 전반에 적용 가능한 범용 텍스트 기반 모델 개발에 기여할 수 있다.
Perturbation landscape 모델링
최근의 세포 표현(cell representation)에 관한 시각은, 세포를 단순히 분자 특징의 정적인 스냅샷(snapshot)으로 표현하는 것에서 벗어나, perturbation(외부 자극, 처리 등)에 대한 세포의 반응까지 포착할 필요가 있다고 강조한다.
Perturbation 데이터셋이 더 풍부해짐에 따라, 향후의 단일세포 트랜스포머 모델들은 다양한 perturbation screen으로도 pretraining될 것으로 기대된다. 이렇게 하면 세포 상태 및 유전자 조절 네트워크(GRN)에 대한 전체적인 이해에 가까워질 수 있다.
만약 생성형 트랜스포머가 단일세포 데이터를 그 정도로 상세하게 이해하게 된다면, 이는 단일세포 데이터를 모사(synthesize)하는 범용 시뮬레이터가 될 수 있다. 예를 들어, 사용자가 지정한 세포 특성에 맞는 오믹스 데이터를 생성하는 데 쓰일 수 있다.
결론
트랜스포머를 단일세포 오믹스에 적용하는 것은 큰 가능성을 지니고 있지만, 철저한 평가 체계가 갖춰지기 전까지는 주의가 필요하다.
현재로서는 트랜스포머가 비순차적(non-sequential) 오믹스 데이터를 모델링하기에 적절한 아키텍처인지, 또는 해당 도메인에서 기존 접근법들보다 우월한 성능을 내는지는 불분명하다.
그럼에도 불구하고, 다른 머신러닝 모델들과 달리 트랜스포머는 대규모의, 다양하고 라벨 없는 데이터로 사전학습되었을 때, 여러 작업에서 성능이 향상되는 특성을 보여왔다.
이처럼 효과적인 self-supervised pretraining은, 단일세포 오믹스 데이터가 앞으로 더 많이 쌓일수록, 트랜스포머가 후속 작업에 걸쳐 적용될 수 있는 잠재력을 가진다. 하지만, 모든 단일세포 생물학을 아우르는 일반적인 트랜스포머(foundation transformer)를 개발하겠다는 목표는 매우 매력적이지만, 아직은 실현되기 매우 먼 이야기다.
트랜스포머가 다른 분야의 다양한 모달리티에서 성공을 거두었지만, 단일세포 오믹스 데이터를 모델링하는 데에는 아직 초기 단계에 머물러 있다.
모델이 학습한 내용을 해석하는 지표(metrics)와 attention score 해석법에 대한 일반적인 연구와 함께, 이 빠르게 성장하는 분야의 발전을 지켜보는 것은 흥미로운 일이 될 것이다.
추가적인 나의 생각
=> XAI를 접목시켜서, 왜 해당 생물학적 결과를 도출했는지를 모델이 설명할 수 있다면 ..?!
코드 넣고 실험해볼 수 있는 논문찾아볼 예정..돌려볼 수 있는 범위하에 +공간전사체학이나 foundation model 관련 논문..
https://theislab.github.io/single-cell-transformer-papers/single-cell-transformers
Transformers in Single-Cell Omics
theislab.github.io


